Downloaded 561 times
















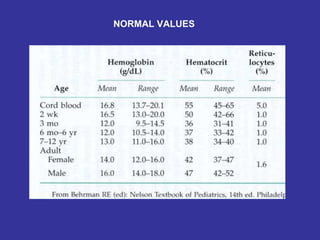




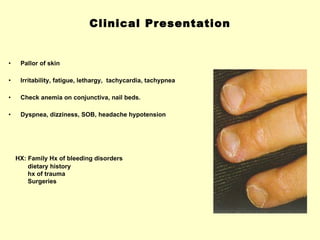
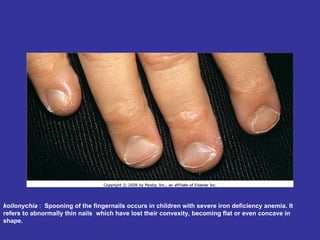
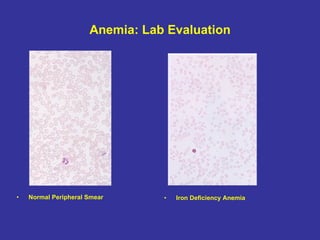


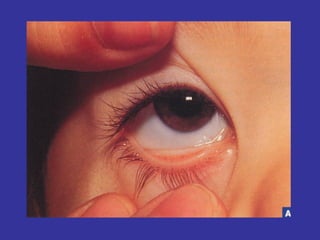



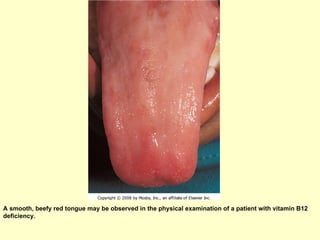



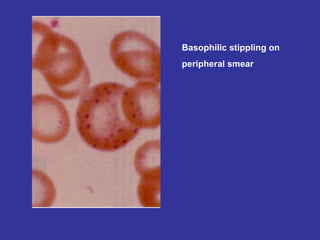








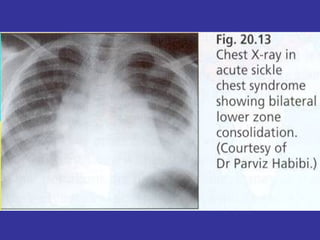
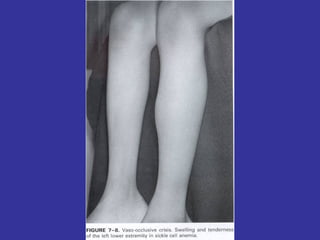
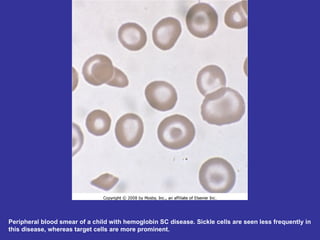
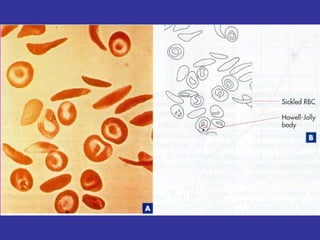
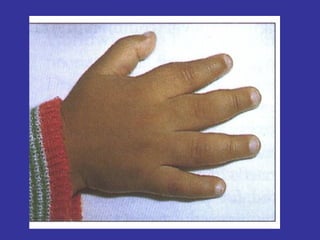
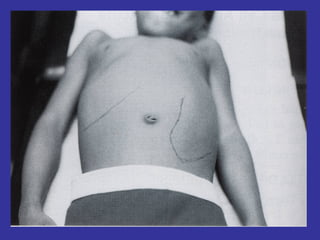




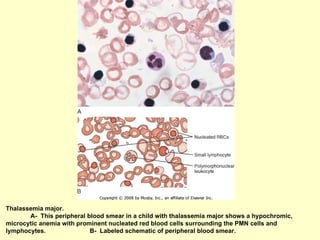
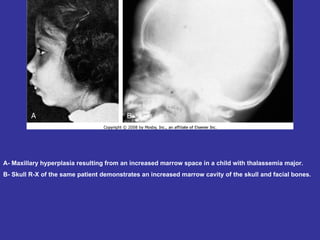














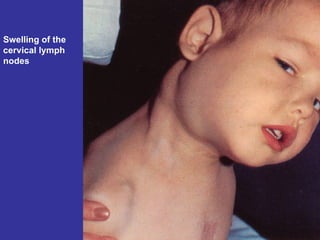
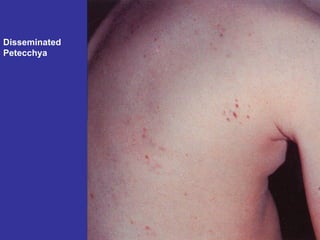
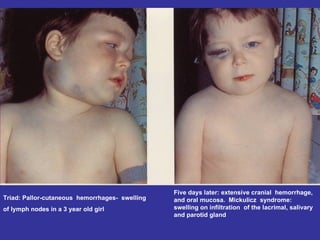




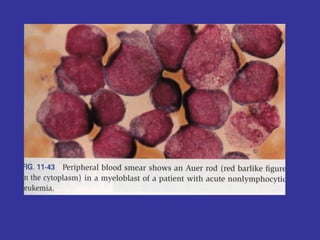









Hematologic markers such as hemoglobin (Hb), hematocrit (Ht), mean corpuscular volume (MCV), and mean corpuscular hemoglobin concentration (MCHC) provide information about red blood cell (RBC) counts, size, and hemoglobin levels. Iron deficiency anemia results in microcytic, hypochromic RBCs and low levels of serum ferritin, iron, and transferrin saturation due to insufficient iron intake or absorption to support normal hemoglobin synthesis. Clinical presentation includes pallor, fatigue, and cardiovascular symptoms. Laboratory evaluation reveals microcytic RBCs on peripheral smear along with low Hb, Ht, MCV, MCH, and iron stores.



























































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)















