Downloaded 5,721 times



![CLASSIFICATION OF HEMOLYTIC ANEMIAS
INTRACORPUSCULAR
DEFECTS
EXTRACORPUSCULAR
FACTORS
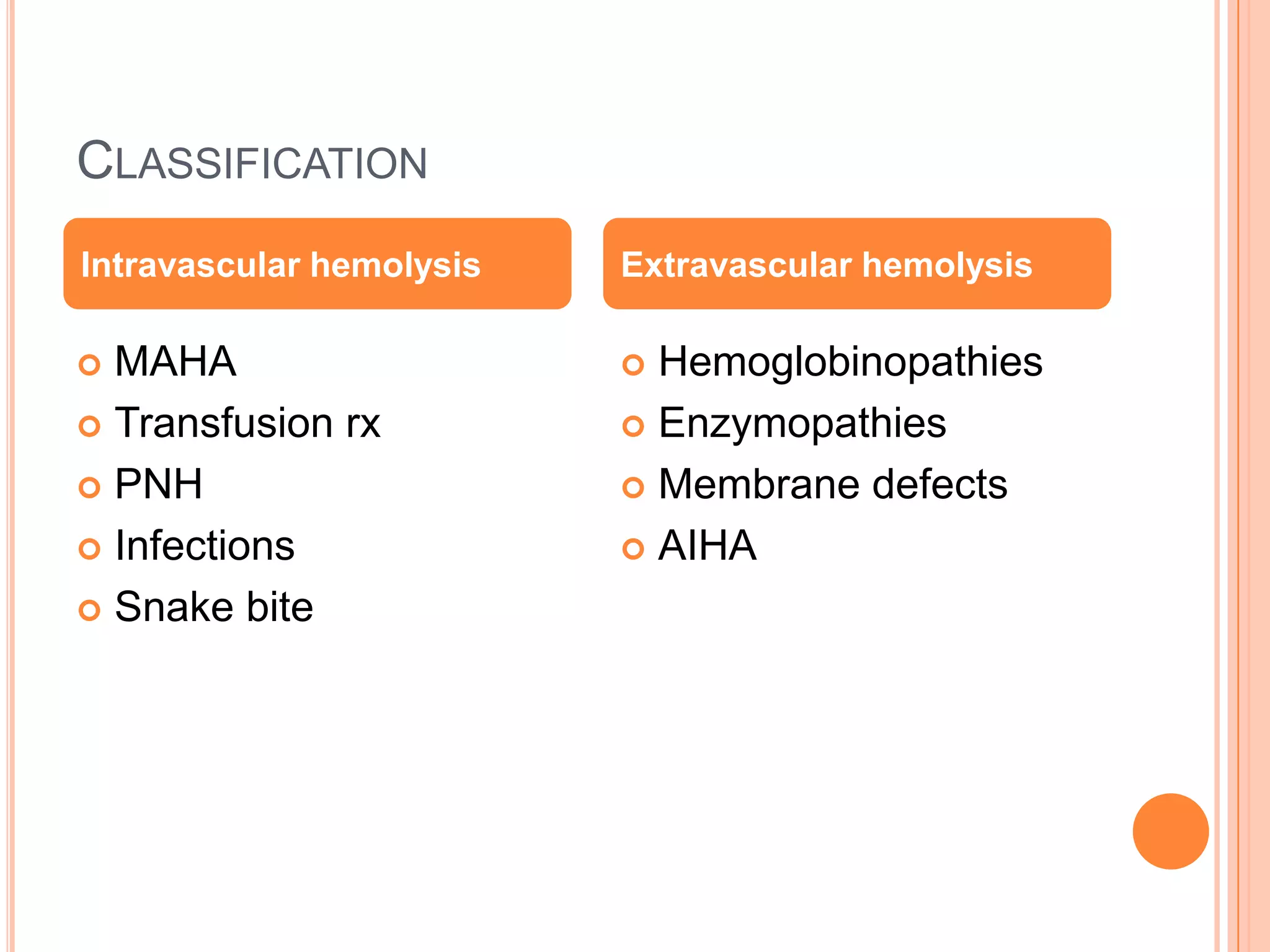
HEREDITARY •HEMOGLOBINOPATHIES
•ENZYMOPATHIES
•MEMBRANE-
CYTOSKELETAL DEFECTS
•FAMILIAL HEMOLYTIC
UREMIC SYNDROME
ACQUIRED •PAROXYSMAL
NOCTURNAL
HEMOGLOBINURIA
•MECHANICAL DESTRUCTION
[MICROANGIOPATHIC]
•TOXIC AGENTS
•DRUGS
•INFECTIOUS
•AUTOIMMUNE](https://image.slidesharecdn.com/approachtohemolyticanemia-131001003025-phpapp02/75/Approach-to-hemolytic-anemia-4-2048.jpg)


![GENERAL FEATURES
OF HEMOLYTIC DISORDERS
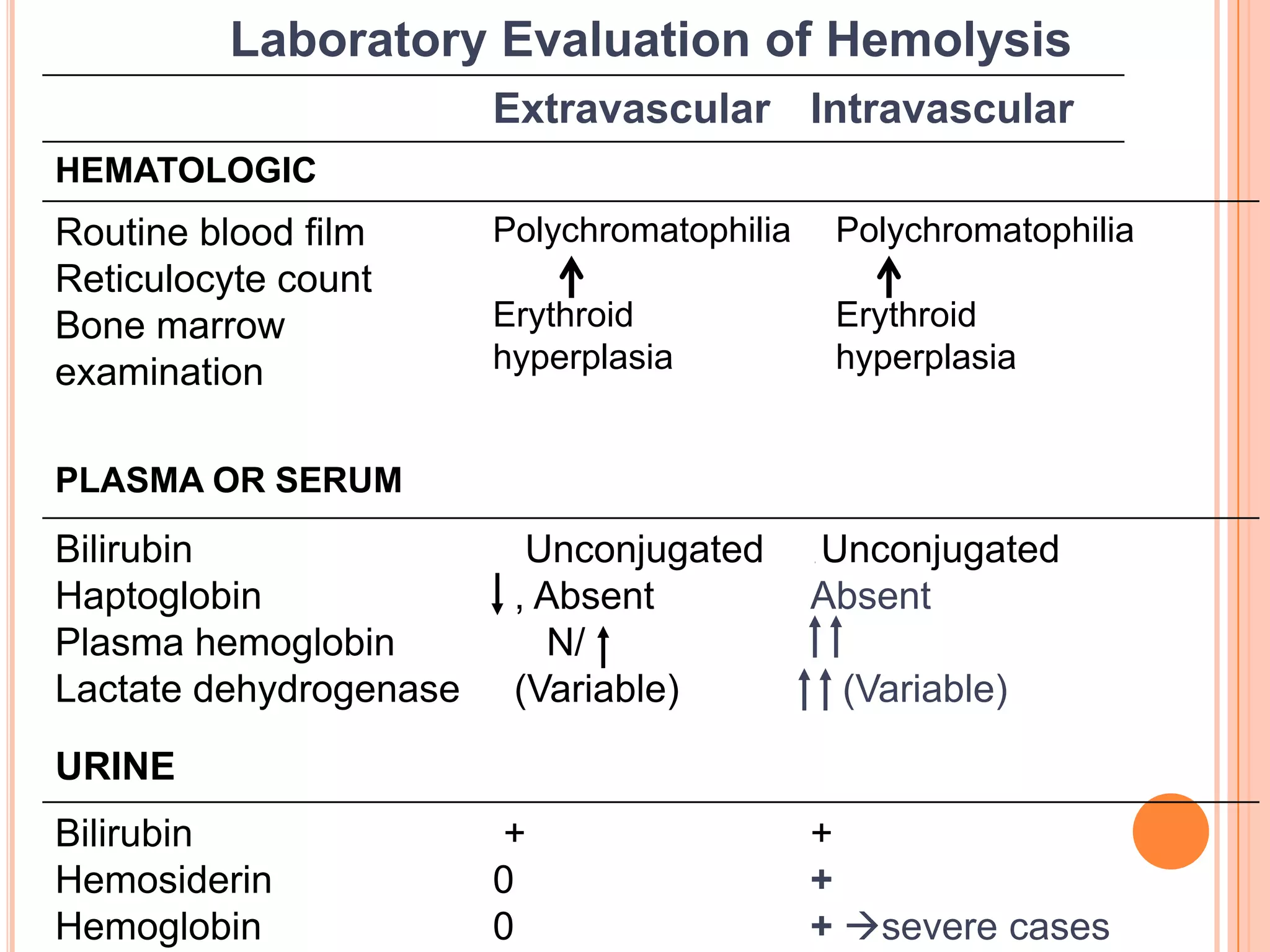
GENERAL EXAMINATION - JAUNDICE, PALLOR
BOSSING OF SKULL
PHYSICAL FINDINGS - ENLARGED SPLEEN
HEMOGLOBIN - FROM NORMAL TO SEVERELY REDUCED
MCV - USUALLY INCREASED
RETICULOCYTES - INCREASED
BILIRUBIN - INCREASED[MOSTLY UNCONJUGATED]
LDH - INCREASED
HAPTOGLOBULIN - REDUCED TO ABSENT](https://image.slidesharecdn.com/approachtohemolyticanemia-131001003025-phpapp02/75/Approach-to-hemolytic-anemia-8-2048.jpg)















![Definitive risk Possible risk Doubtful risk
antimalarials Primaquine
Dapsone
cholrproguanil
chloroquine quinine
Sulphonamides/
sulphones
Sulphametoxazole
Dapsone
Sulfasalazine
Sulfadimidine
Sulfisoxazole
Sulfadiazine
Antibacterials/
Antibiotics
Cotrimoxazole
Nalidixic acid
Nitrofurantoin
Ciprofloxacin
Norfloxacin
Cholramphenicol
p-Aminosalicylic
acid
Antipyretic/
Analgesics
Acetanilide
Phenazopyridine
[pyridium]
Acetylsalicylic acid
High dose[>3g/d]
Acetylsalicylic acid
[<3g/d]
Acetaminophen](https://image.slidesharecdn.com/approachtohemolyticanemia-131001003025-phpapp02/75/Approach-to-hemolytic-anemia-36-2048.jpg)

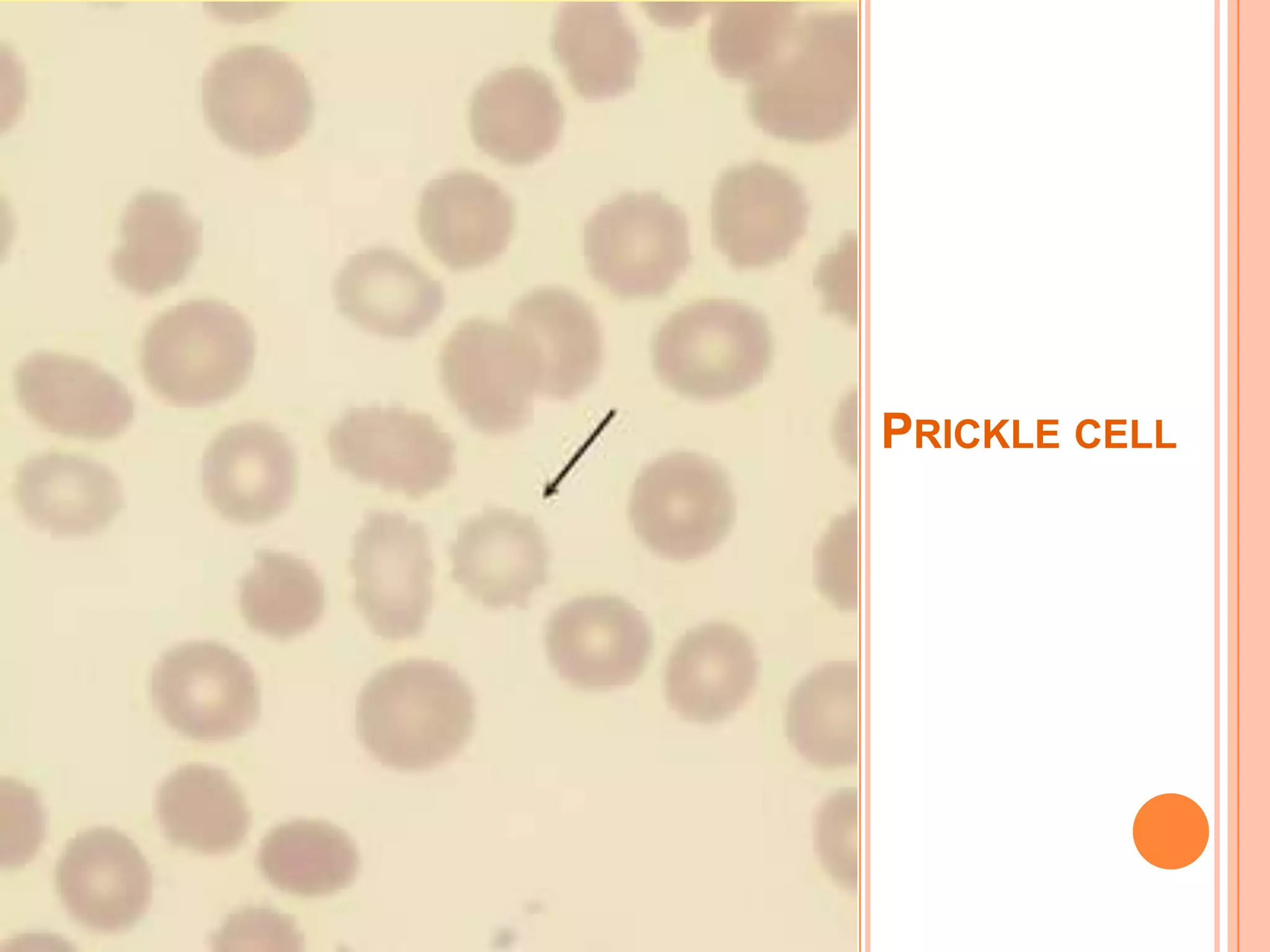

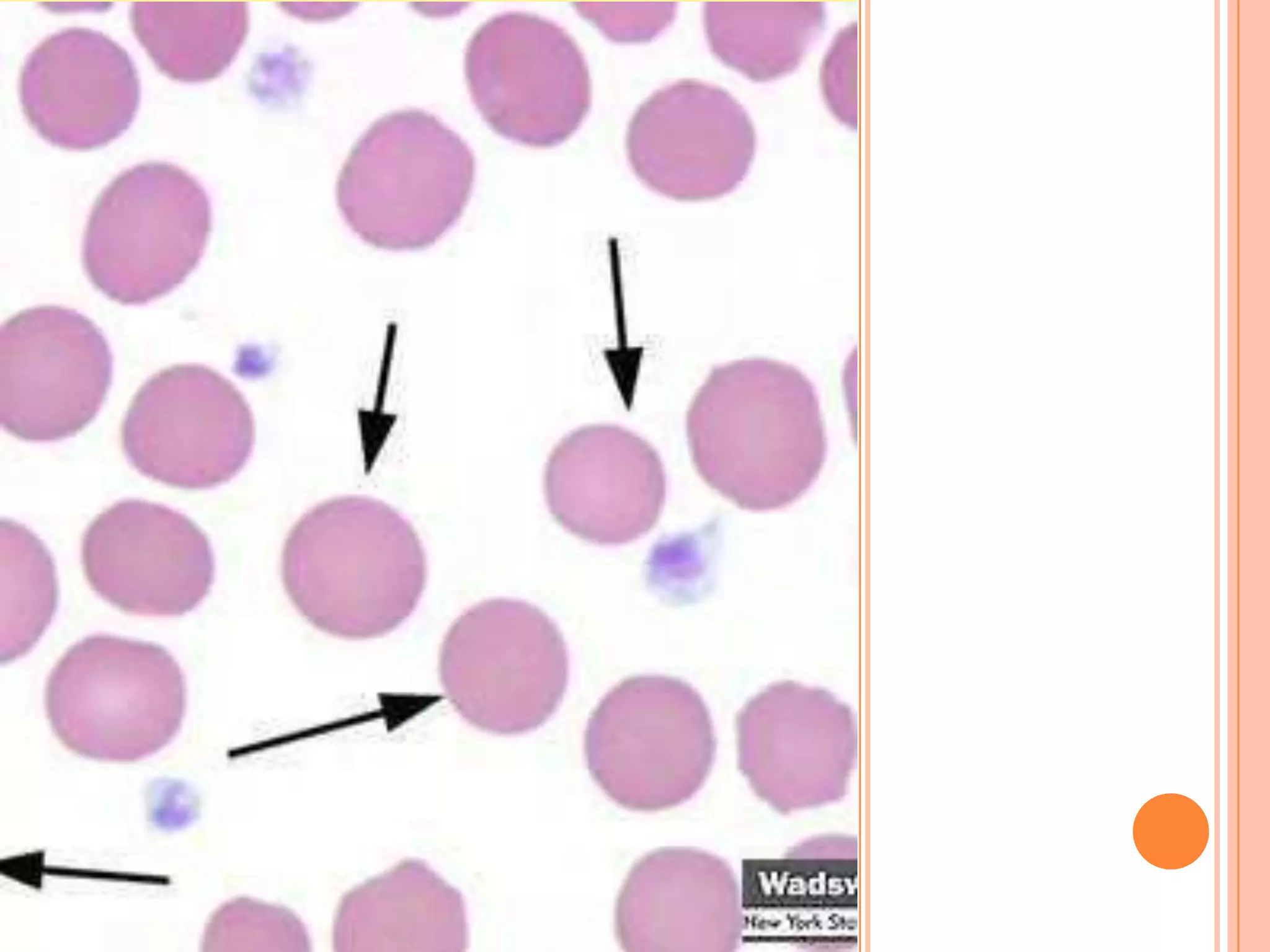








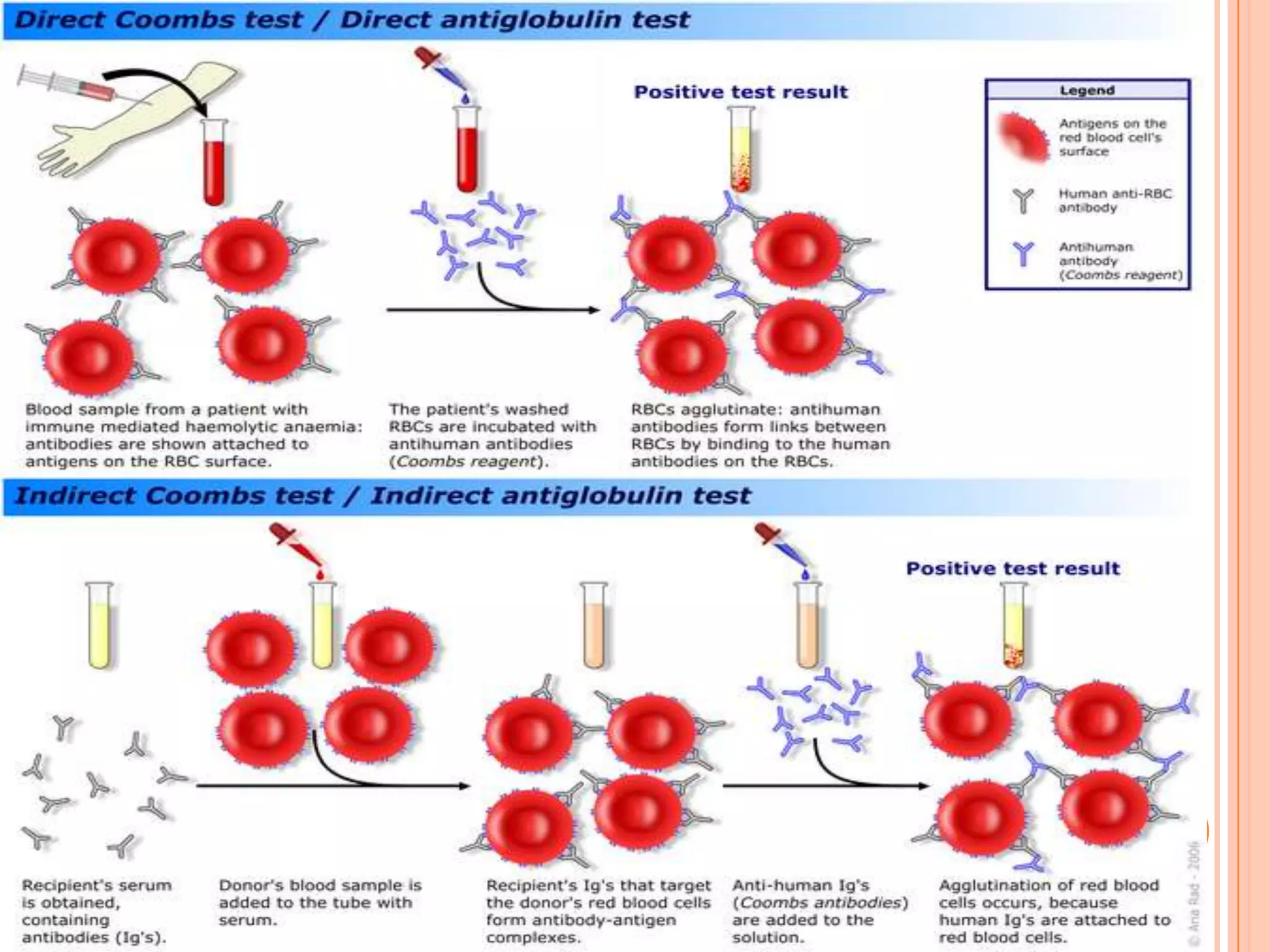






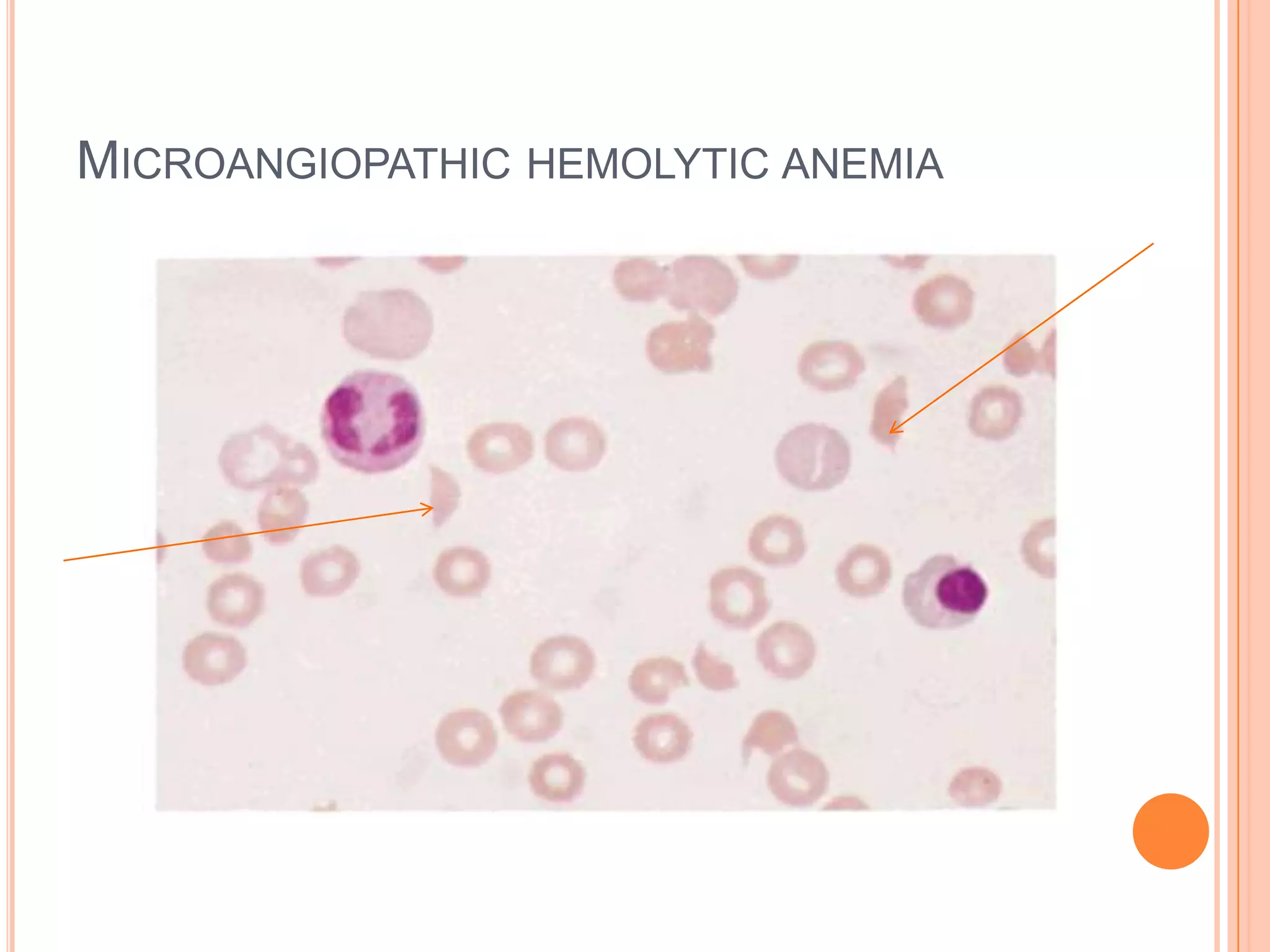



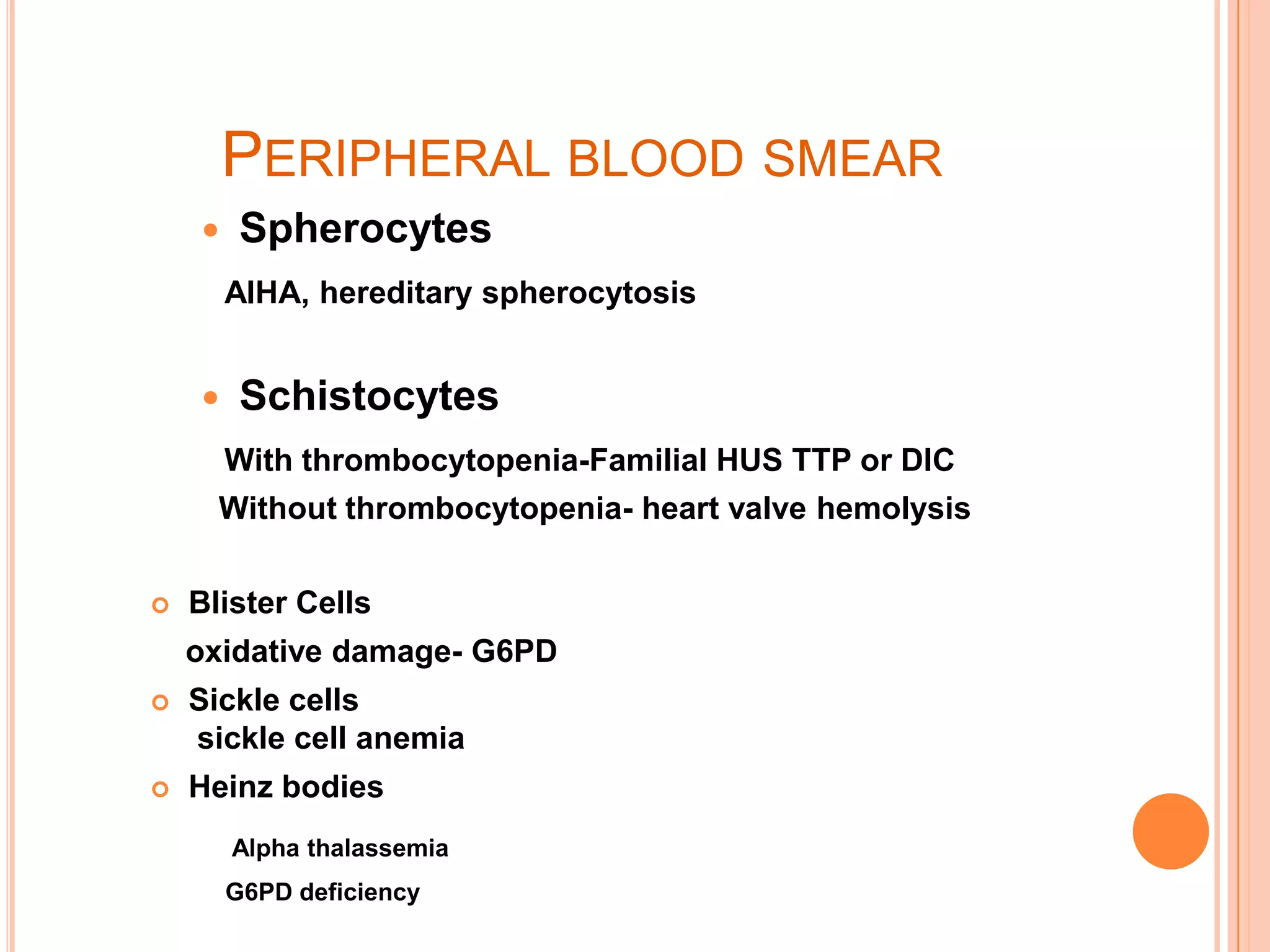
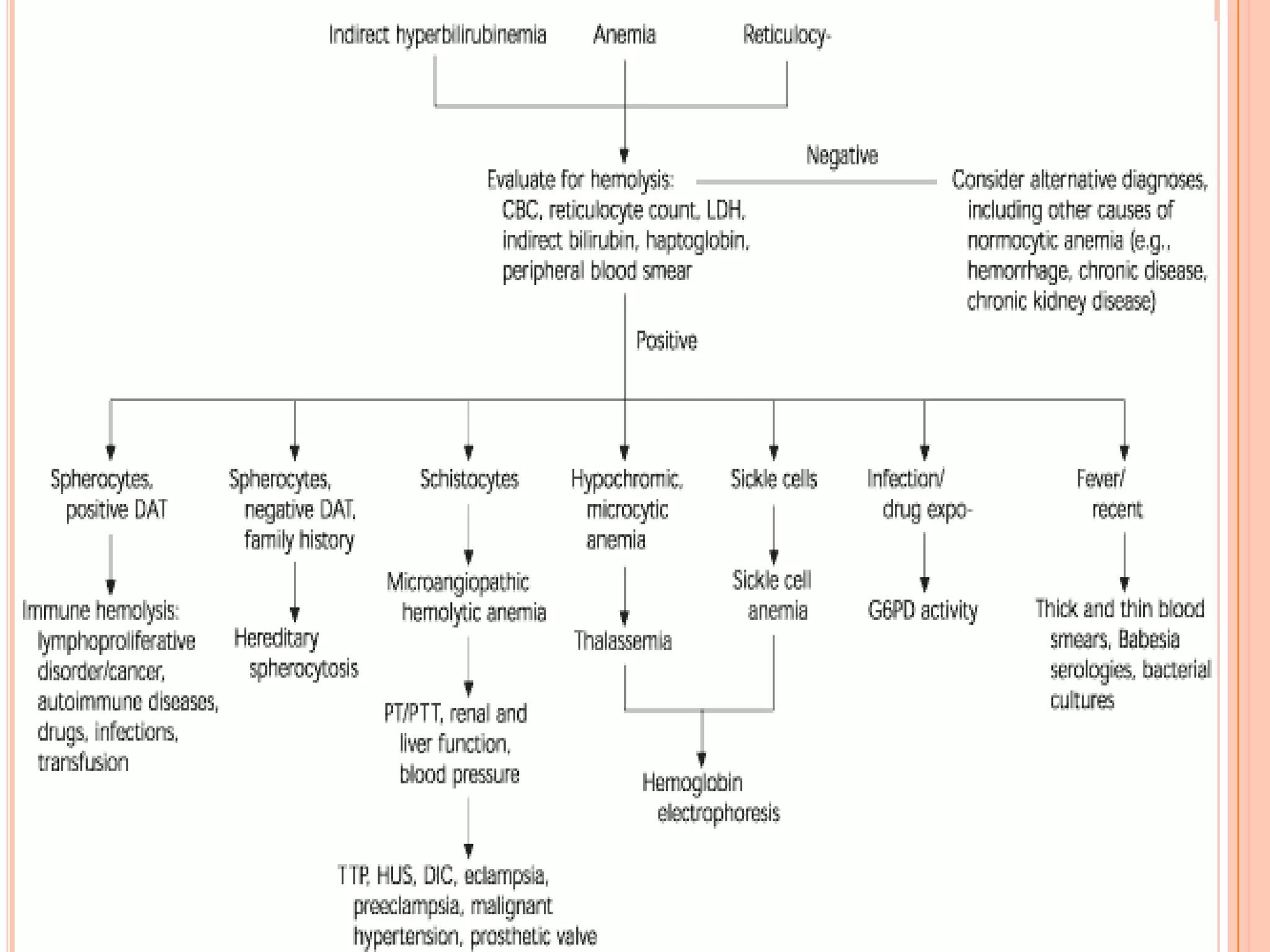



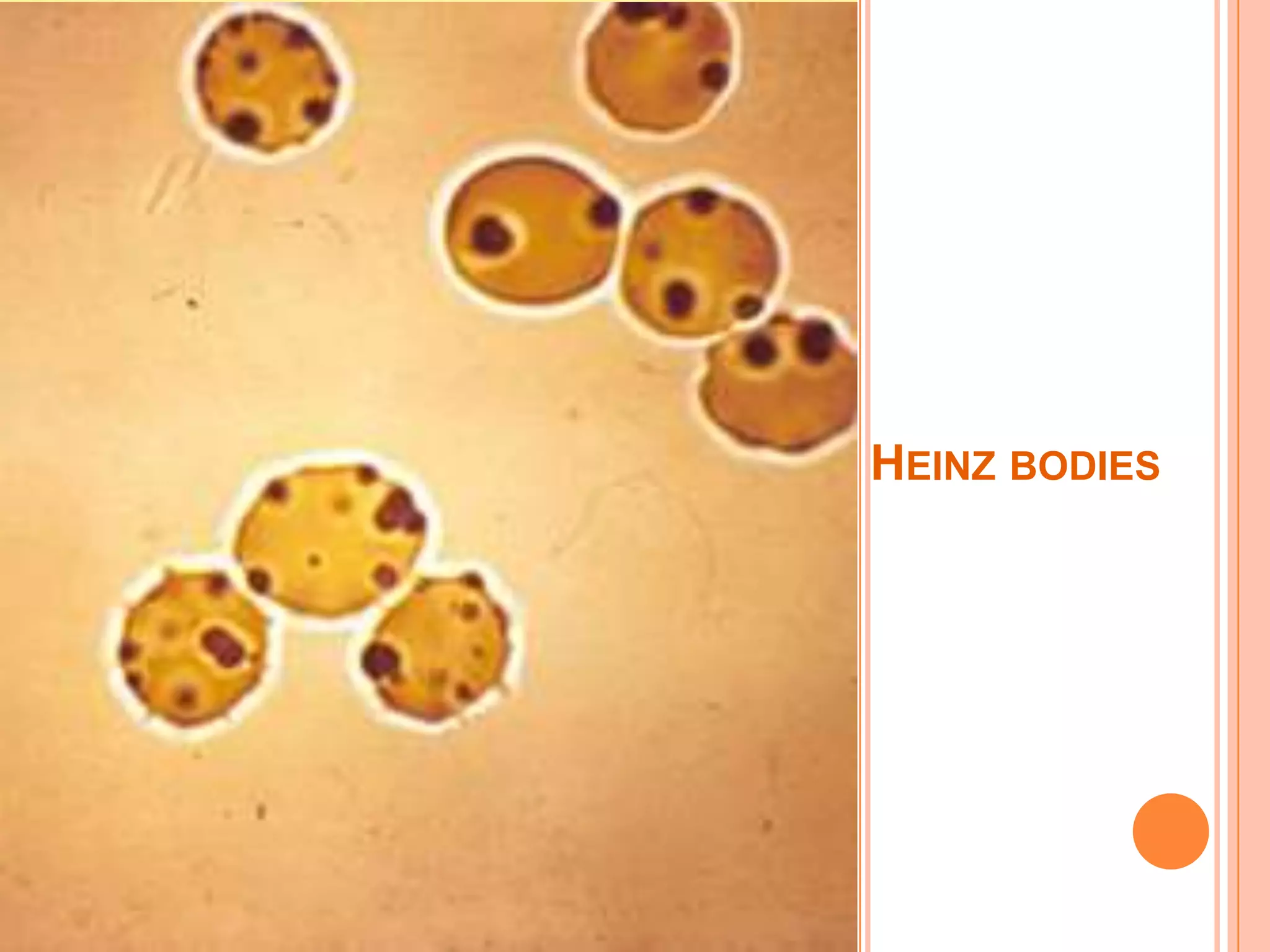
Dr Sarath Menon presents an approach to diagnosing and classifying hemolytic anemia. Hemolytic anemia results from increased red blood cell destruction and bone marrow compensation. It can be congenital/hereditary or acquired. Classification includes intracorpuscular defects like hemoglobinopathies and enzymopathies, and extracorpuscular factors like mechanical destruction, toxic agents, infections, and autoimmune causes. Diagnosis involves confirming hemolysis and determining the etiology through history, physical exam, peripheral smear, and ancillary lab tests. Common etiologies discussed in detail include sickle cell disease, thalassemia, G6PD deficiency, membrane defects like hereditary spherocytosis, and autoimmune




















































































