Thalassemia
Dr. Kalpana Malla
MD Pediatrics
Manipal Teaching Hospital
Download more documents and slide shows on The Medical Post [ www.themedicalpost.net ]
2.
AKA
• VON JAKSCHANEMIA
• COOLEY’S ANEMIA
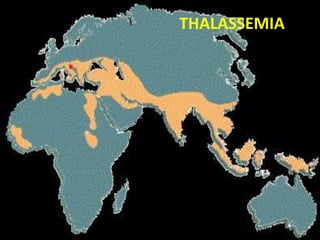
• “THALASSA” : GREEK WORD - GREAT SEA
– first observed - MEDITTERANIAN SEA
DEFINTION
• Thalassemia sydromesare a
heterogenous group of inherited anemias
characterised by reduced or absent
synthesis of either alpha or Beta globin
chains of Hb A
• Most common single gene disorder
5.
BASICS - 3types of Hb
1. Hb A - 2 α and 2 β chains forming a tetramer
• 97% adult Hb
• Postnatal life Hb A replaces Hb F by 6 months
2. Fetal Hb – 2α and 2γ chains
• 1% of adult Hb
• 70-90% at term. Falls to 25% by 1st month and
progressively
3. Hb A2 – Consists of 2 α and 2 δ chains
• 1.5 – 3.0% of adult Hb
6.
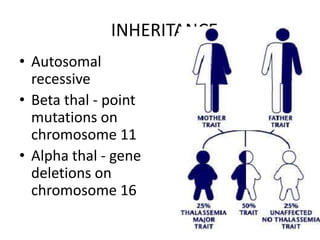
INHERITANCE
• Autosomal
recessive
• Beta thal - point
mutations on
chromosome 11
• Alpha thal - gene
deletions on
chromosome 16
7.
Classification
• If synthesisof α chain is suppressed – level of all
3 normal Hb A (2α ,2β),A2 (2α ,2 δ),F(2α ,2γ)
reduced – alpha thalassemia
• If β chain is suppressed - adult Hb is suppressed
- beta thalassemia
8.
CLASSIFICATION
• α-thalassemia
Hb H (β4)
Hb-Bart’s ( 4)
• β-thalassemia
• β+ thal : reduced synthesis of β globin chain,
heterozygous
• β 0 thal : absent synthesis of β globin chain,
homozygous------ Hb A - absent
Hb F (α2 2)
Hb A2 (α2 δ2)
9.
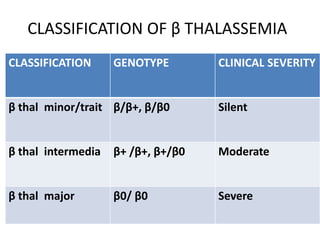
CLASSIFICATION OF βTHALASSEMIA
CLASSIFICATION GENOTYPE CLINICAL SEVERITY
β thal minor/trait β/β+, β/β0 Silent
β thal intermedia β+ /β+, β+/β0 Moderate
β thal major β0/ β0 Severe
PATHOPHYSIOLOGY
• Since ẞchain synthesis reduced -
1. gamma 2 and delta δ2 chain combines with
normally produced α chains ( Hb F (α2 2) , Hb A2
(α2 δ2) - Increased production of Hb F and Hb A2
2. Relative excess of α chains → α tetramers forms
aggregates →precipitate in red cells → inclusion
bodies → premature destruction of maturing
erythroblasts within the marrow (Ineffective
erythropoiesis) or in the periphery
(Hemolysis)→ destroyed in spleen
14.
PATHOPHYSIOLOGY
Anemia result fromlack of adequate Hb A
→ tissue hypoxia→↑EPO production →
↑ erythropoiesis in the marrow and
sometimes extramedullary → expansion
of medullary cavity of various bones
Liver spleen enlarge → extramedullay
hematopoiesis
15.
EFFECTS OF MARROWEXPANSION
• Pathological fractures due to cortical thinning
• Deformities of skull and face
• Sinus and middle ear infection due to
ineffective drainage
• Folate deficiency
• Hypermetabolic state -> fever, wasting
• Increased absorption of iron from intestine
16.
HEPATOMEGALY
• Extra medullaryerythropoeisis
• Iron released from breakdown of
endogenous or transfused RBCs cannot be
utilized for Hb synthesis – hemosiderosis
• Hemochromatosis
• Infections – transfusion related - Hep B,C,
HIV
• Chronic active hepatitis
17.
SPLENOMEGALY
• Extra medullaryhematopoeisis
• Work hypertrophy due to constant
hemolysis
• Hypersplenism (progressive
splenomegaly)
INFECTIONS -CAUSES
• Poornutrition
• Increased iron in body
• Blockage of monocyte-macrophage
system
• Hypersplenism- leukopenia
• Infections associated with transfusions
20.
ACCUMULATION OF IRON
•Deposition in pituitary - endocrine
disturbance - short stature, delayed puberty,
poor sec. sexual characteristics
• Hemochromatosis - cirrhosis of liver
• Cardiomyopathy (cardiac hemosiderosis) -
cardiac failure, sterile pericarditis, arrythmias,
heart block
• Deposition in pancreas -diabetes mellitus
21.
ACCUMULATION OF IRON
• Lungs: restrictive lung defects
• Adrenal insufficiency
• Hypothyroidism, hypoparathyroidism
• Increased susceptibity to infections (iron
favours bacterial growth) espc : Yersinia
infections
22.
CLINICAL FEATURES (THALMAJOR)
INFANTS:
• Age of presentation: 6-9 mo (Hb F replaced by
Hb A)
• Progressive pallor and jaundice
• Cardiac failure
• Failure to thrive, gross motor delay
• Feeding problems
• Bouts of fever and diarrhea
• Hepatosplenomegaly
23.
CLINICAL FEATURES (THALMAJOR)
BY CHILDHOOD:
Growth retardation
Severe anemia-cardiac dilatation
Transfusion dependant
Icterus
Changes in skeletal system
24.
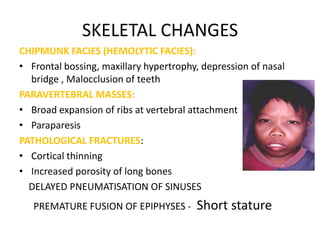
SKELETAL CHANGES
CHIPMUNK FACIES(HEMOLYTIC FACIES):
• Frontal bossing, maxillary hypertrophy, depression of nasal
bridge , Malocclusion of teeth
PARAVERTEBRAL MASSES:
• Broad expansion of ribs at vertebral attachment
• Paraparesis
PATHOLOGICAL FRACTURES:
• Cortical thinning
• Increased porosity of long bones
DELAYED PNEUMATISATION OF SINUSES
PREMATURE FUSION OF EPIPHYSES - Short stature
25.
Others
• Delayed menarche
• Gall-stones, leg ulcers
• Pericarditis
• Diabetes/ cirrhosis of liver
• Evidence of hypersplenism
26.
CLINICAL FEATURES (THAL
INTERMEDIA)
• Moderate pallor, usually maintains Hb >6gm%
• Anemia worsens with pregnancy and
infections (erythroid stress)
• Less transfusion dependant
• Skeletal changes present, progressive
splenomegaly
• Growth retardation
• Longer survival than Thal major
27.
CLINICAL FEATURES (THALMINOR)
• Usually ASYMPTOMATIC
• Mild pallor, no jaundice
• No growth retardation, no skeletal
abnormalities, no splenomegaly
• MAY PRESENT AS IRON DEFICIENCY ANEMIA
(Hypochromic microcytic anemia)
• Unresponsive/ refractory to Fe therapy
• Normal life expectancy
28.
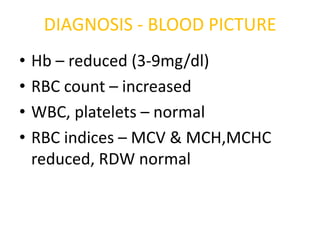
DIAGNOSIS - BLOODPICTURE
• Hb – reduced (3-9mg/dl)
• RBC count – increased
• WBC, platelets – normal
• RBC indices – MCV & MCH,MCHC
reduced, RDW normal
29.
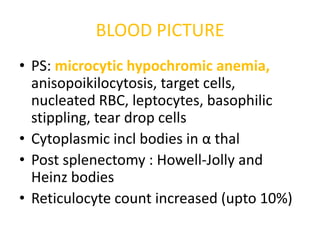
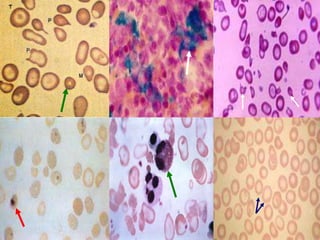
BLOOD PICTURE
• PS:microcytic hypochromic anemia,
anisopoikilocytosis, target cells,
nucleated RBC, leptocytes, basophilic
stippling, tear drop cells
• Cytoplasmic incl bodies in α thal
• Post splenectomy : Howell-Jolly and
Heinz bodies
• Reticulocyte count increased (upto 10%)
31.
DIAGNOSIS
• Osmotic fragilitytest : increased- resistance
to h’lysis
• T. bilirubin, I. bilirubin – increased
• Haptoglobulin and hemopexin – depleted
• S. Fe, ferritin elevated, Transferrin –
saturated
• B.M. study: hyperplastic erythropoesis
32.
DIAGNOSIS
• Red cell survival – decreased using
• Folate levels- concurrently decreased
• Free erythrocyte porphyrin - normal
• Serum uric acid-raised
• Haemosiderinuria
33.
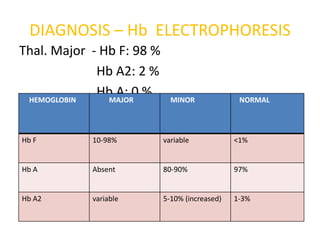
DIAGNOSIS – HbELECTROPHORESIS
Thal. Major - Hb F: 98 %
Hb A2: 2 %
HEMOGLOBIN
Hb A: 0 %
MAJOR MINOR NORMAL
Hb F 10-98% variable <1%
Hb A Absent 80-90% 97%
Hb A2 variable 5-10% (increased) 1-3%
34.
Radiological changes
• Smallbones (hand ) – earliest bony change,
rectangular appearance,medullary portion of
bone is widened &bony cortex thinned out
with coarse trabecular pattern in medulla
• Skull – widened diploid spaces – interrupted
porosity gives hair on end appearance
• Delayed pneumatization of sinuses – maxilla
appears overgrown with prominent malar
eminences
35.
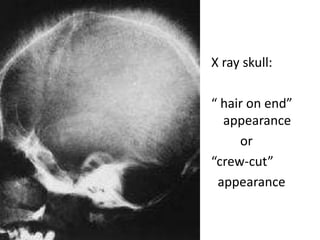
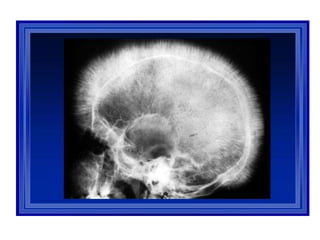
X ray skull:
“hair on end”
appearance
or
“crew-cut”
appearance
37.
IRON OVERLOAD ASSESSMENT
• S. Ferritin
• Urinary Fe excretion
• Liver biopsy
• Chemical analysis of tissue Fe
• Endomyocardial biopsies
• Myocardial MRI indexes
• Ventricular function – ECHO, ECG
38.
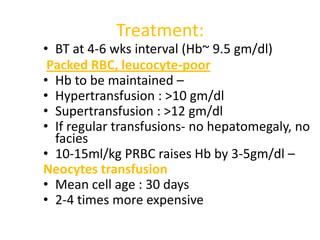
Treatment:
• BT at4-6 wks interval (Hb~ 9.5 gm/dl)
Packed RBC, leucocyte-poor
• Hb to be maintained –
• Hypertransfusion : >10 gm/dl
• Supertransfusion : >12 gm/dl
• If regular transfusions- no hepatomegaly, no
facies
• 10-15ml/kg PRBC raises Hb by 3-5gm/dl –
Neocytes transfusion
• Mean cell age : 30 days
• 2-4 times more expensive
39.
CHELATION THERAPY -DESFERRIOXAMINE
• ( 1 unit of blood contains 250 mg iron)
• Iron-chelating agents: desferrioxamine-
• Dose: 30-60mg/kg/day
• IV / s/c infusion pump over 12 hr period 5-6
days /wk
• Start when ferritin >1000ng/ml
• Best >5 yrs
• Vitamin C 200 mg on day of chelation -
enhances DFO induced urinary excretion of Fe
CHELATION THERAPY- DEFERIPRONE
•Oral chelator - > 2yrs old Dose: 50-100mg/kg/day
• Adverse effects:
Reversible arthropathy
Drug induced lupus
Agranulocytosis
• Other oral chelators
Deferrothiocine
Pyridoxine hydrazine
ICL-670 – removes Fe from myocardial cells
42.
TREATMENT - SPLENECTOMY
•Deferred as long as possible. At least till 5-6
yrs age
• Splenectomy (indications):
• Massive splenomegaly causing
mechanical discomfort
• Progressively increasing blood
transfusion requirements (>180-200
ml/kg/yr) packed RBC
43.
BONE MARROW TRANSPLANTATION
•BEST METHOD FOR CURE
• Risk factors:
Hepatomegaly >2cm
Portal fibrosis
Iron overload
Older age
44.
Newer therapies:
• GENEMANIPULATION AND REPLACEMENT
• Remove defective β gene and stimulate γ gene
• 5-azacytidine increases γ gene synthesis
• Hb F AUGEMENTATION
• Hydroxyurea
• Myelaran
• Butyrate derivatives
• Erythropoetin in Thal intermedia
45.
OTHER SUPPORTIVE MEASURES
•Tea – thebaine and tannins– chelate iron
• Vitamin C – increases iron excretion
• Restrict Fe intake – decrease meat, liver, spinach
• Folate – 1 mg/day
• Genetic counselling
• Psychological support
• Hormonal therapy – GH, estrogen, testosterone,
L-thyroxine
• Treatment of CCF
α-thalassemia:
• Deletion onalpha globin locus on Chr 16
• Defective synthesis of α-globin chain
• Excess of - chains - in the fetus (Hb Bart- 4)
Excess of β-chains in the adult (Hb H- β4)
ALPHA THALASSEMIA
• Highestprevalence in Thailand
• α chains shared by fetal as well as adult life.
Hence manifests both times
• These thalassemias don’t have ineffective
erythropoesis because β and γ are soluble chains
and hence not destroyed always
• α Thalassemia trait mimics Fe deficiency anemia
• Silent carrier – silent – not identified
hematologically, diagnosed when progeny has Hb
Barts/ Hb H
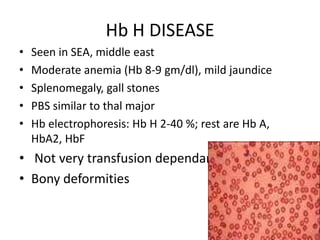
Hb H DISEASE
• Seen in SEA, middle east
• Moderate anemia (Hb 8-9 gm/dl), mild jaundice
• Splenomegaly, gall stones
• PBS similar to thal major
• Hb electrophoresis: Hb H 2-40 %; rest are Hb A,
HbA2, HbF
• Not very transfusion dependant
• Bony deformities
56.
Hb BARTS
• HbBarts has γ4, then later in infancy β4
• Severe hypoxia as Hb Barts has high affinity for
oxygen
57.
Haemoglobin Bart’s:
• Mostsevere manifestation of alpha thalassemia
• Hydrops fetalis – Fatal unless intrauterine transfusions
• Stillborn or die within a few hours
• Severe anemia , edematous, mildly jaundiced,
ascites, hepatosplenomegaly, cardiac failure
• Looks like Rh incompatilibity
• Increased incidence of toxemia
of pregnancy
58.
• DIAGNOSIS
• Hbelectrophoresis:
80-90 % Hb Bart’s
Hb H
Hb Portland
No Hb A, Hb A2 or Hb F
• Treatment: immediate exchange transfusion
59.
DIAGNOSIS OF αTHALASSEMIA
• CBC, PS, BM study
• Heinz bodies in HbH disease – brilliant cresyl
blue
• Hb electrophoresis – for HbH and Hb Barts
• α/β chain ratio decreased
60.
Treatment:
• Generally notreqd
• Blood transfusion , iron chelation therapy –
For transfusion dependent cases
• Avoidance of oxidant drugs
• Prompt treatment of infections
• Folic acid supplementation
• Splenectomy
• BM transplantation, gene therapy
61.
Thank you
Download moredocuments and slide shows on The
Medical Post [ www.themedicalpost.net ]

![Thalassemia
Dr. Kalpana Malla
MD Pediatrics
Manipal Teaching Hospital
Download more documents and slide shows on The Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/hematology-thalassemia-120108093334-phpapp01/85/Thalassemia-1-320.jpg)











































![Thank you
Download more documents and slide shows on The
Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/hematology-thalassemia-120108093334-phpapp01/85/Thalassemia-61-320.jpg)











































































































