Downloaded 980 times







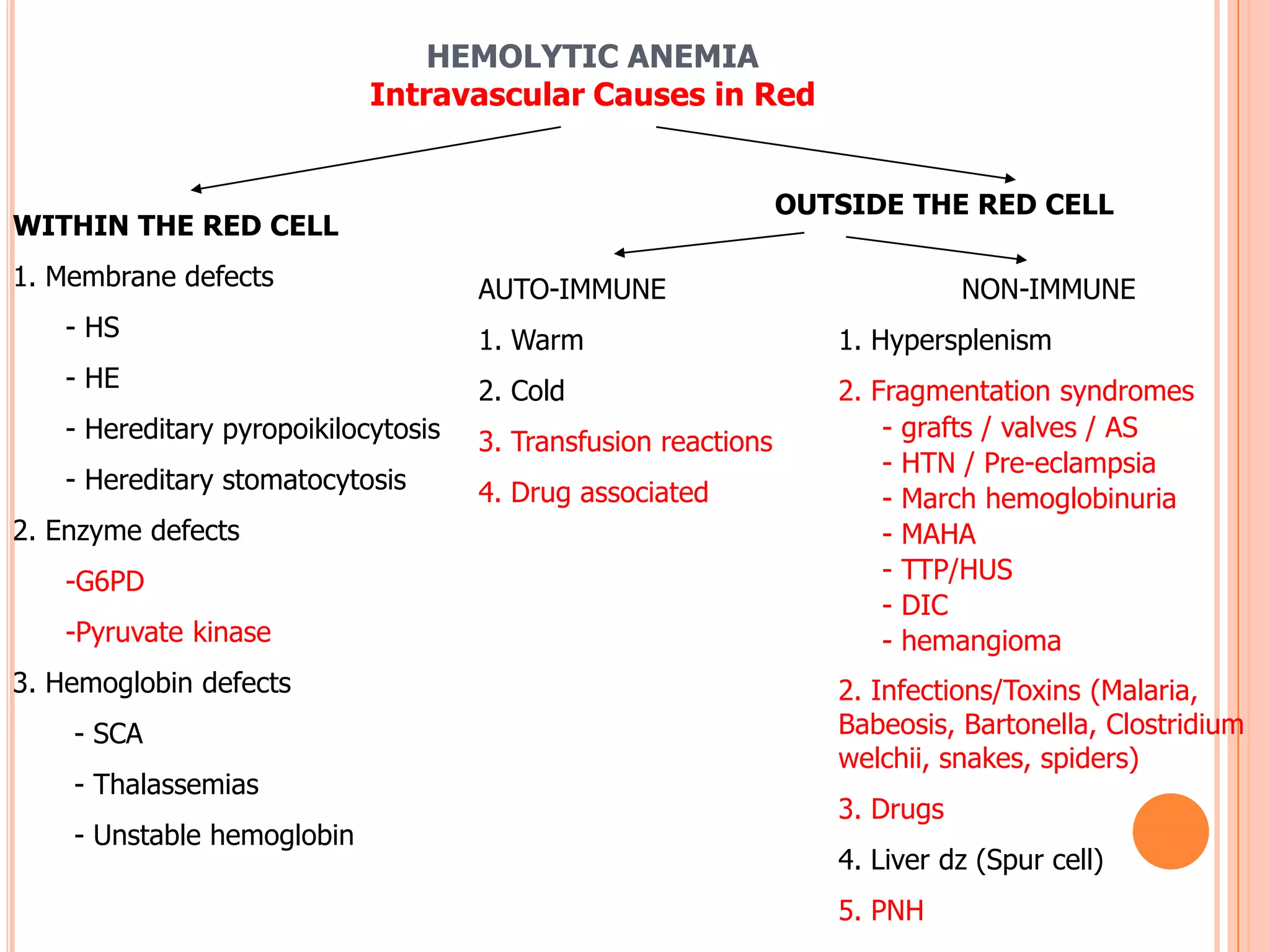



































This document provides a classification and overview of hemolytic anemia. It discusses intracorpuscular defects like hereditary membrane defects (spherocytosis, elliptocytosis), enzyme defects (G6PD, pyruvate kinase), and hemoglobinopathies. Extracorpuscular defects include immune hemolytic anemia (autoimmune, alloimmune) and nonimmune causes. Evaluation of anemia involves hematological parameters. Thalassemias are classified based on affected globin chain (alpha, beta). Common hereditary spherocytosis causes premature RBC destruction and can be treated with splenectomy. G6PD deficiency results in drug-induced hemolysis.





































































































