DEFINITION
The musculardystrophies are a heterogeneous group of inherited disorders
characterized by progressive weakness and degeneration of skeletal muscles.
The muscular dystrophies (MD) are genetically determined progressive
disorders of muscle characterized by cycles of muscle fiber necrosis,
regeneration, eventual fibrosis and replacement with fatty tissue.
Muscular dystrophy is a group of diseases that cause progressive weakness and
loss of muscle mass. In muscular dystrophy, abnormal genes (mutations)
interfere with the production of proteins (Dystrophin) needed to form healthy
muscle.
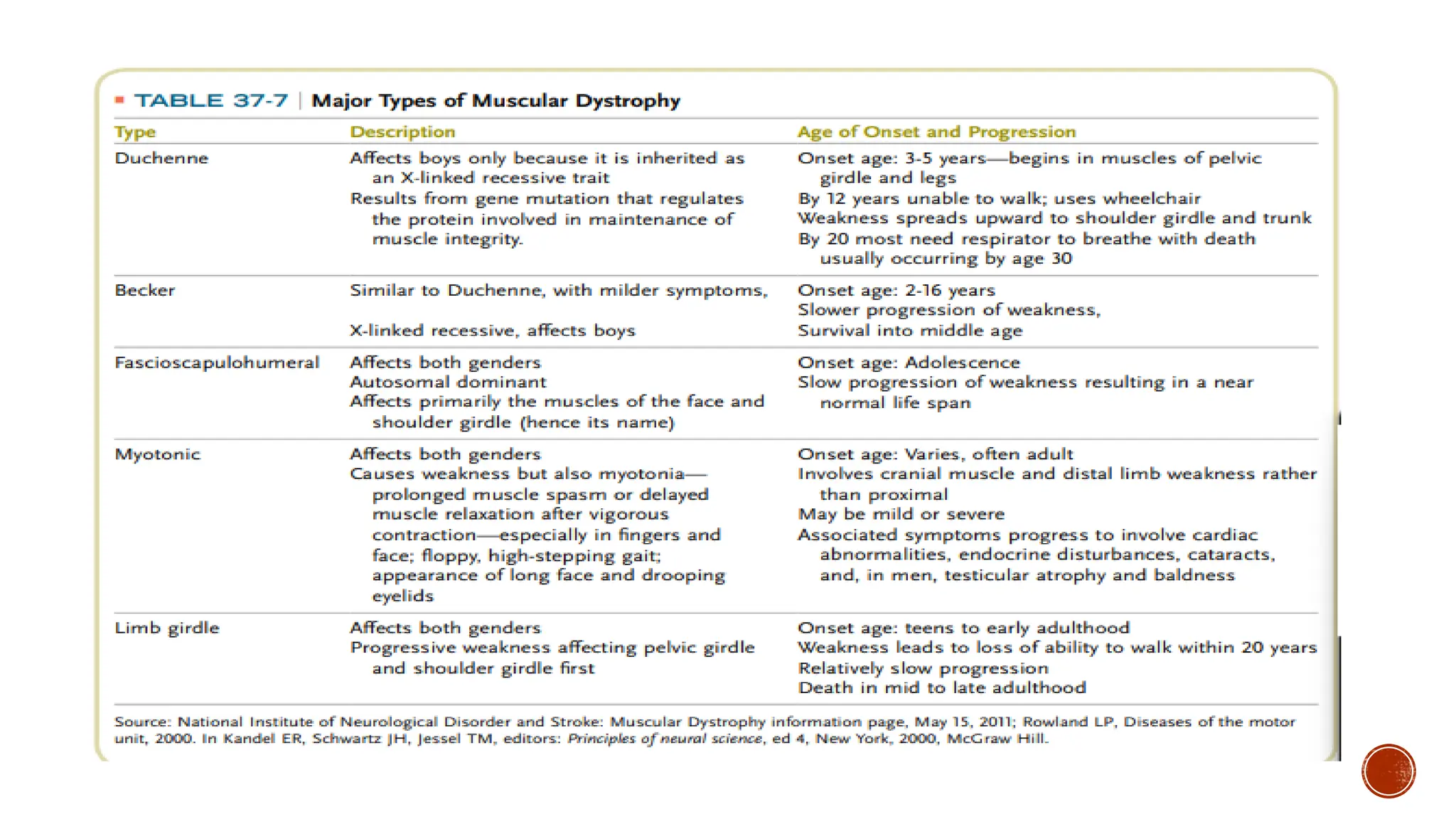
DUCHENNE MUSCULAR DYSTROPHY
Duchenne muscular dystrophy, the most common form of MD, is an X-linked disorder (i.e,
associated with a gene on the X chromosome) that was first described over a century ago.
Duchenne muscular dystrophy is characterized by progressive wasting of skeletal muscles,
with the limb-girdle muscles first showing weakness by the age of 5 years, followed by an
inability to walk by the ages of 8 to 12 years.
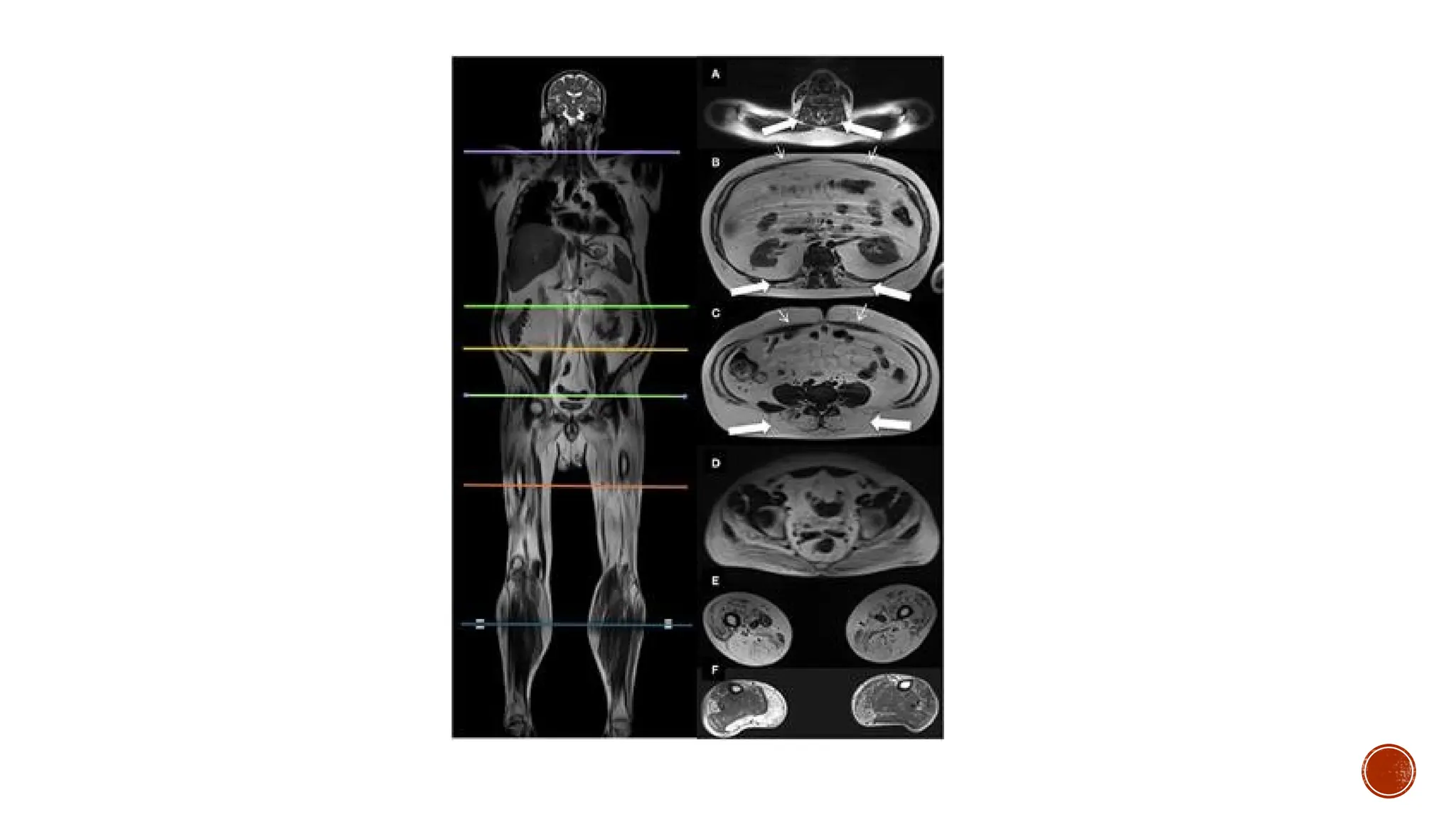
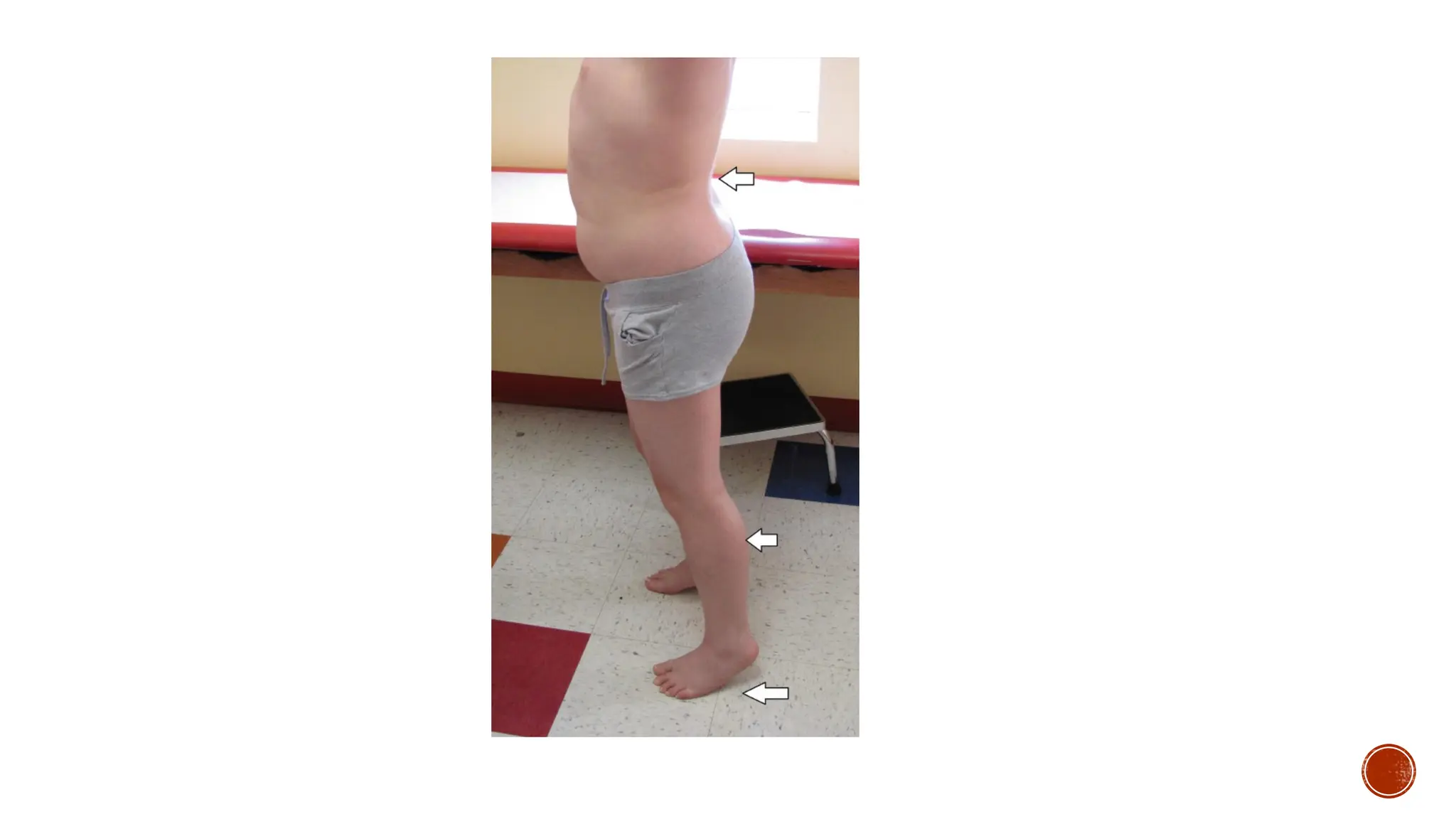
Other findings include elevated creatine kinase levels, pseudo hypertrophic calf muscles,
decreased levels of activity, and, in some patients, cognitive impairment. At the cellular level,
pathological changes include the absence of dystrophin at the membrane of the muscle fibers,
increased adipose and connective tissue between muscle fibers, increased variability in muscle
fiber size, infiltration of inflammatory cells, and centrally located nuclei, which are indicative
of degenerating and regenerating muscle fibers.
6.
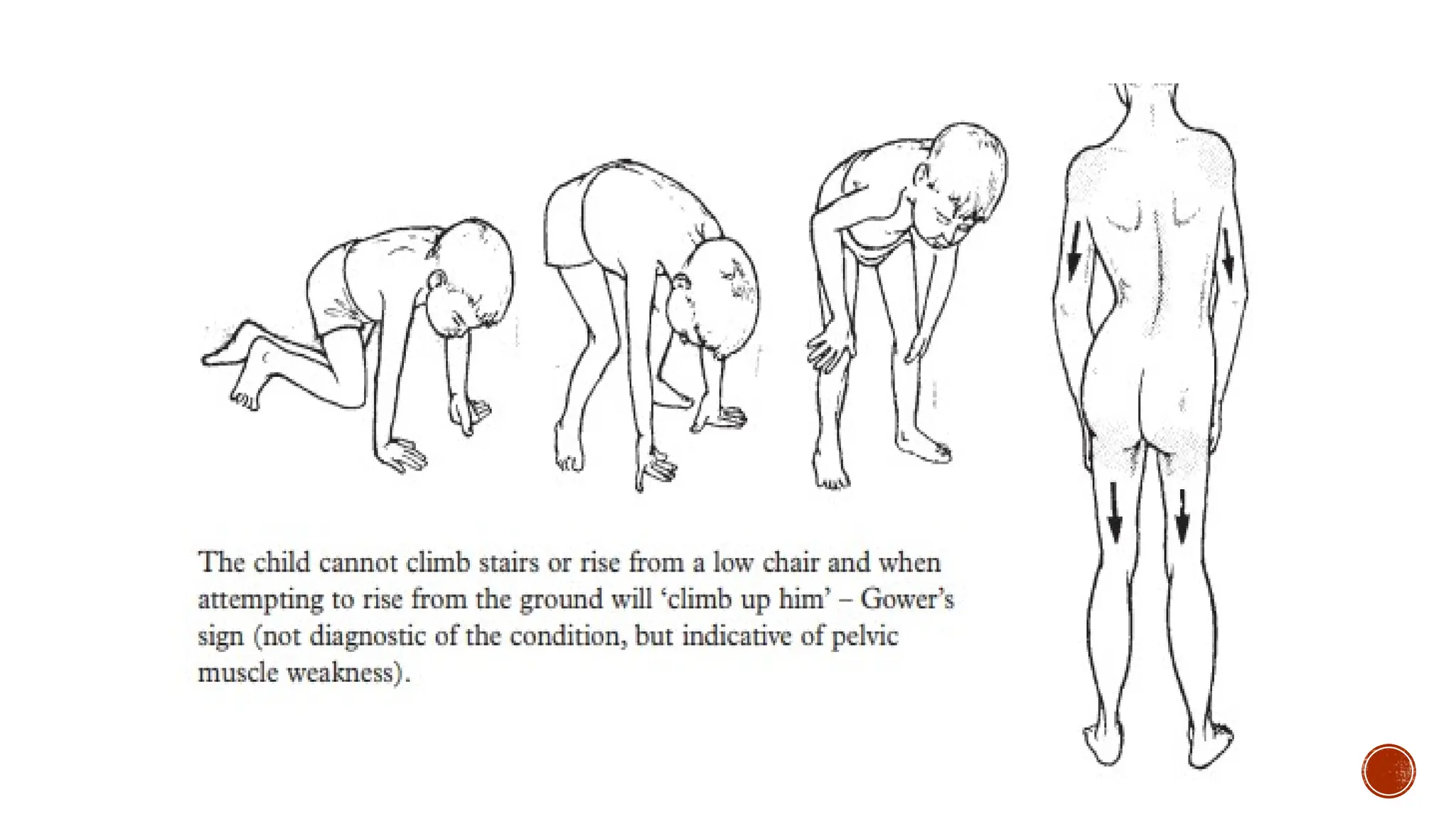
Parents typicallydo not seek medical care early on, because children with DMD look
“normal” for the first few years of life. Between the ages of 2 and 5 years, they begin to show
signs of clumsiness, falling, and gait changes, as well as difficulty ascending stairs. By age 6
years, the child often develops contractures of the calf muscles and an exaggerated lordosis of
the spine. By this time, the child has a positive Gowers’ sign, and the loss of strength (the
ability of a muscle to produce force) progresses throughout the upper body and lower
body. The scoliosis often becomes severe, producing secondary pulmonary complications and
requiring surgical fusion to stop its progress. Death usually occurs in the second or third
decade of life due to cardiac or respiratory impairment.
Duchenne muscular dystrophy is caused by the absence of dystrophin, a 427 kDa protein
found on the cytoplasmic surface of the plasma membrane of muscle fibers (the sarcolemma)
in skeletal and cardiac muscle
Approximately 1 in 3,500 newborn males worldwide are affected with DMD.
7.
BECKER MUSCULAR DYSTROPHY
Abnormalities within the dystrophin gene may be associated with a spectrum of
presentations from Duchenne to the milder condition described by Becker.
Becker MD is rarer than Duchenne MD – incidence 1:35000, presenting at a later
age usually with limb girdle involvement and pseudohypertrophy. Cardiac
involvement may be symptomatic in up to 10% of affected individuals and female
carriers and is not related to the mutation or the severity of limb muscle disease.
The onset of BMD is usually between the ages of 5 and 15 years, but can occur as late as the
fourth decade of life. The phenotypic presentation of BMD is similar to that of DMD, but is
clinically milder and with more variability and a much slower progression. Patients with BMD
do not have contractures or severe scoliosis, and many live well into adulthood, sometimes to
a normal life span.
8.
LIMB GIRDLE MUSCULARDYSTROPHY
As their name implies, these myopathies are characterized by weakness of the proximal muscles
in the upper and lower extremities. Onset can occur in childhood and the clinical presentation
can mimic DMD, but onset more often occurs in late adolescence or early adulthood.
Some of the most severe forms of LGMD present at birth, falling into the category of congenital
muscular dystrophy (CMD). The heart is usually not affected, but patients with LGMD should be
screened routinely because some will develop cardiomyopathy.
Limb-girdle muscular dystrophies can either be autosomal dominant (single gene defect on a
chromosome from either parent or one copy of a mutant gene and one normal gene, known as
type 1 LGMD) or autosomal recessive (a defect or mutation on the gene from the chromosome
of each parent is needed, known as type 2 LGMD). The type 2 LGMDs are more severe, with
some resembling DMD in severity.
The majority of LGMDs are autosomal recessive. Patients exhibit a variable severity of muscle
disease, usually involving scapular winging and weakness of proximal limb and trunk muscles.
9.
FACIOSCAPULOHUMERAL MD
AfterDMD and LGMDs, facioscapulohumeral muscular dystrophy (FSHD) is the third most
common inherited muscle disease.
Incidence 1–2:100000.The mechanism by which this mutation causes disease is not
known.
The clinical features include:
– Facial weakness (which may be mild or asymmetrical)
– Periscapular weakness producing winging of the scapula and rising up of the
scapulae on attempted abduction
– Weakness of the humeral muscles
– A predominantly proximal lower limb pattern of weakness giving a dromedary or
camel backed gait.
Pseudohypertrophy is not a feature.
10.
Severity isvariable, ranging from severe childhood forms to later onset disease
that may be asymptomatic.
CK levels may only be raised to 1.5–2 upper limit or normal.
EMG and muscle biopsy will show myopathic abnormalities but have no specific
features; although secondary inflammatory change on biopsy may lead to an
erroneous diagnosis of Polymyositis.
Cardiac involvement is not a feature.
High frequency sensorineural hearing loss and exudative retinal telangiectasis
complicate some early onset cases (Coat’s syndrome). Prognosis is dependent on
the degree of respiratory muscle involvement. Some may benefit from ventilatory
support.
11.
MYOTONIC DYSTROPHY
Myotonicdystrophies are the most common form of MD in adults. Myotonic dystrophies are
now recognized as genetically heterogeneous diseases, caused by 2 distinct mutations.
Myotonic dystrophy type 1 (DM1) is caused by an expansion of a CTG trinucleotide repeat in
a gene for an enzyme.
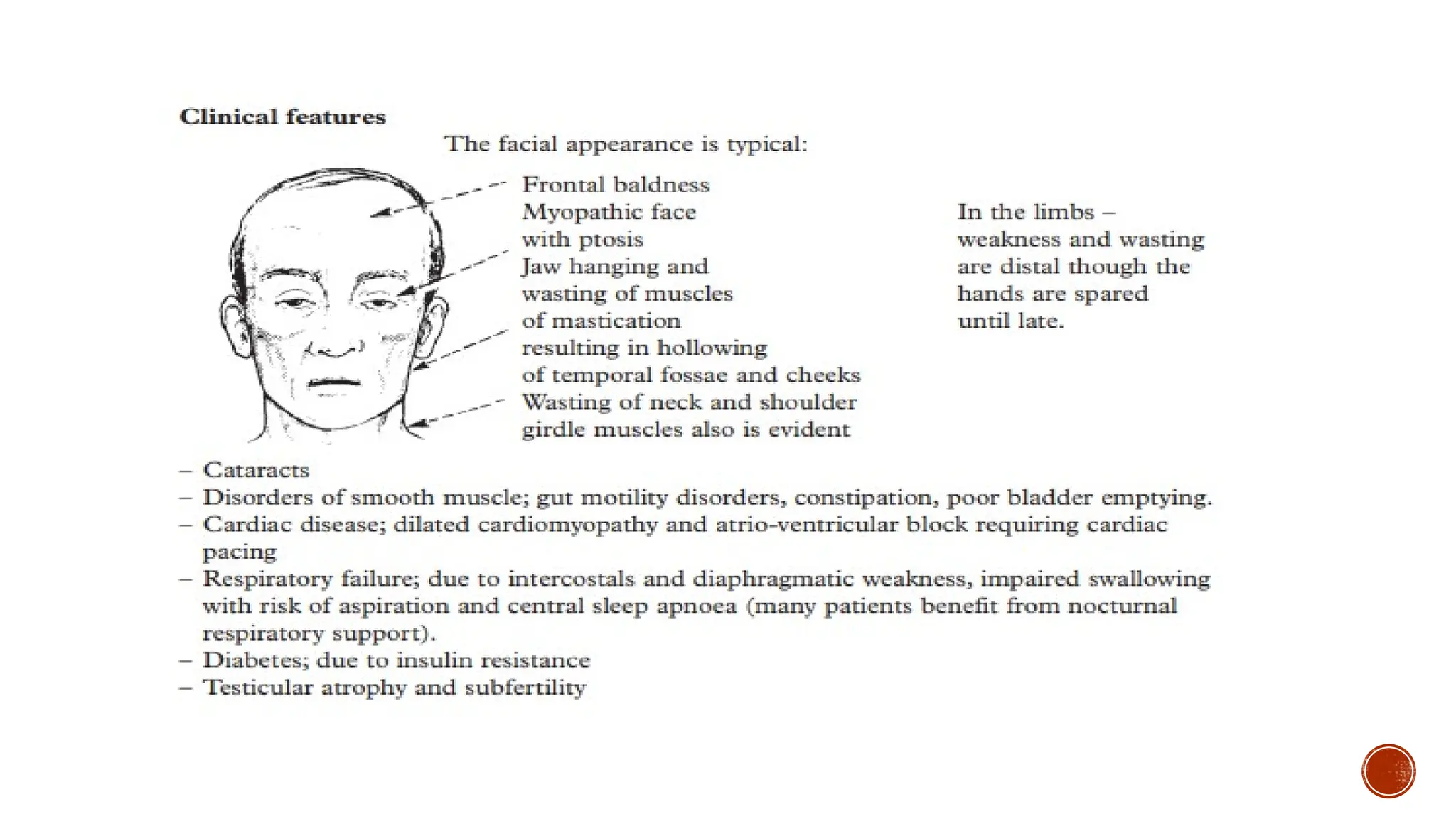
The features of classic myotonic dystrophy include myotonia (skeletal muscle
hyperexcitability and slowed relaxation time), slowly progressive muscle weakness, cardiac
conduction defects, pain, peripheral neuropathy, frontal balding, temporal wasting, cataracts,
and endocrine disturbances such as diabetes. Myotonic dystrophy type 1 may present in
infancy (CMD) or childhood, but the disorder typically presents in young adults, with a
prevalence estimated to be as high as 1/8,000.
Myotonic dystrophy type 2 (DM2) is caused by an expansion of a CCTG repeat in intron 1 of
the gene ZNF9, which encodes zinc finger protein 9 on chromosome 3q. Both DM1 and DM2
are inherited in an autosomal dominant fashion, and both affect multiple organ systems.
12.
Whilst neuromuscularfeatures may not be prominent, the condition is usually
characterized by the presence of MYOTONIA – failure of immediate muscle
relaxation after contraction has ceased.
14.
EMERY- DREIFUSS MUSCULARDYSTROPHY
Rare but important because of its cardiac complications. Both X-linked and
dominant forms reported (the dominant form is now classified as LGMD type 2).
Contractures of the spine produce an appearance of hyperextension. Contractures
of elbows and ankles occur early.Weakness may be in a scapuloperoneal
distribution. Life threatening cardiac condition defects are virtually universal and
ventricular tachyarrhythmias occur in a proportion. Patients will require pacing
and some have implanted defibrillators. Respiratory muscle weakness may occur.
Emery-Dreifuss muscular dystrophy presents clinically with the triad of early contractures,
muscle weakness, and cardiac conduction defects. Weakness occurs in the shoulder girdle and
distal lower extremities (“humeroperoneal” weakness) and usually starts in childhood,
although symptoms can begin at any time between the neonatal period and the third decade.
Contractures are usually out of proportion to the weakness and affect many joints, especially
the elbows, followed by the ankles and cervical spine. Although patients with EDMD are not
wheelchair-bound, contractures are a major cause of morbidity, creating more functional
impairment than the weakness. The major cause of mortality is cardiac disease, which often
results in premature and sudden death.
15.
OCCULOPHARYNGEAL MUSCULAR DYSTROPHY
This is another very rare pattern of weakness associated with a small GCG
trinucleotide expansion in the PABP2 gene on chromosome 14. Inheritance is
autosomal dominant. Occurs with a mean age of onset of 50 years with a
combination of ptosis, ophthalmoparesis and dysphagia. Limb weakness may
occur. Muscle biopsy shows rimmed vacuoles and filamentous intranuclear
inclusions.
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant disorder that is
characterized by progressive eyelid ptosis and progressive dysphagia, followed by
involvement of other muscles of the head and neck, and eventually proximal limb
weakness. The extraocular muscles are usually spared, although not always.76
Onset occurs in
late adulthood, and a clinical test that is sometimes used is timed swallowing, which is 2 times
slower than normal in people with OPMD.
16.
CONGENITAL MUSCULAR DYSTROPHY
Congenital muscular dystrophies are a class of relatively rare conditions that present in
infancy. Because of the vagaries of the naming system, many forms of CMD are classified
with the limb-girdle muscular dystrophies (e.g., severe congenital autosomal recessive
muscular dystrophy [SCARMD]). The typical CMD cases are often those associated with
disturbances in the central nervous system. Newborns and infants with CMD have significant
weakness and up to a 10-fold increase in the blood level of the enzyme creatine kinase, a
general indicator of muscle damage. Clinical manifestations include muscle weakness,
hypotonia, delayed motor development, and severe contractures with consequent joint
deformities.
17.
DISTAL MUSCULAR DYSTROPHY
This group of rare diseases affects adult men and women. It causes weakness and
wasting of the distal muscles of the forearms, hands, lower legs and feet. It is
generally less severe, progresses more slowly and affects fewer muscles than other
forms of muscular dystrophy.
19.
MEDICAL MANAGEMENT
Becausethis group of diseases is degenerative, the decline of muscle function
cannot be prevented. As yet, there is no cure. Medical management is largely
supportive.“Drug therapy includes corticosteroids to slow muscle degeneration,
anticonvulsants to control seizures and some muscle activity, immunosuppressants
to delay some damage to dying muscle cells, and antibiotics to fight respiratory
infections” Gene therapy is being investigated but is not yet available.
Rehabilitation measures are vital in delaying deformity and achieving maximal
function within the limits of the disease and its debilitating effects. In addition to
occupational therapy, rehabilitation may include physical therapy, speech therapy,
and respiratory therapy services.
Because dystrophin is the central component of a large complex of proteins at the cell
membrane that is missing in DMD, an ideal treatment would be simply to replace the missing
protein. Much of the focus in DMD is on gene therapy to do just that, but delivery of the
dystrophin gene to all muscles of the body has presented some serious challenges.
REHAB TEAM
Acomprehensive team evaluation of the client’s abilities and disabilities should be
administered.
The team includes
the physician, occupational therapist, physical therapist and psychologist.
A social worker may advise the family on community resources.
25.
OT ASSESSMENT
The occupationaltherapist assesses
The client’s functional status in ADLs and IADLs
ROM exercises
Muscle strength
As well as the fit and proper use of adaptive equipment, engagement in leisure
activities and emotional status.
27.
SCALES
Visual analoguescale- to check pain
MRC SCALE- to check muscle strength
Jebson's hand function test
GMFCS- to check gross motor function
BERG BALANCE SCALE- to check balance
BARTHEL INDEX- to check ADL
VFIM- to check ADL in case of children
COPM- Canadian Occupational Performance Scale
OT GOALS
Toimprove the child’s ROM, balance and coordination.
To improve the muscle strength.
To reduce pain and stiffness.
To improve their ADL.
To improve their communication and social skills.
To minimize cardiac issues.
The primary goal of OT is to help the client with MD maintain maximal independence as
long as possible.
To improve Selfcare activities and assistive devices that promote independence during
home, school, leisure, and work activities are a vital part of the treatment program.
Leisure occupations need to be considered in a comprehensive OT intervention plan to
ensure balance in all areas of the client’s life.
Play and laughter are particularly important to the health, well-being, and social
adjustment of the growing youth
30.
ROLE OF OT
After the evaluation process is complete, an OT intervention plan is developed to
address specific concerns.
Motor skills training
Fatigue maintenance and endurance training
Functional mobility
ADL and IADL performance
Leisure interests
Assistive technology needs
31.
TREATMENT
PREVENTING CONTRACTURES:
Stretchingexercises and postural changing to maintain functional performance
skills
Stretching to the most contracture prone muscle groups
AFOs at night to supplement
Joint protection technique
CARDIO:
Breathing exercise
Relaxation techniques
Energy conservation technique
Work simplification technique
32.
ADDRESSSING STRENGTH ANDENDURANCE:
Maintain or improve muscle strength and maximize functional ability
Avoid muscular damage by overwork or injury
Voluntary active exercises such as swimming/ hydrotherapy/ cycling in
ambulatory children currently recommended.
Active exercises may be helpful in maintaining strength, but overexertion and
fatigue must be prevented. Because people with MD may experience cardiac
complications, occupational therapists must be aware of the client’s medical
history and use exercise judiciously, observing cardiac precautions when
necessary. Incorporating exercise into meaningful, age-related activities that
are monitored by a therapist can promote engagement in participation.
Parachute games, obstacle courses, and swimming may be implemented with
frequent built-in rest breaks.
33.
MOBILITY:
Passive stretchingand splinting
Wheelchair and crutches
Walking orthoses- AFOs KAFO
Standing frames, standing wheelchairs and walkers
Transboard
Eventually need indoor lift, van with lift, roll-in shower
Splinting and therapy to prevent hand contractures
Leg braces
Night splints
34.
DYSPHAGIA MANAGEMENT:
Volume &texture modification:
Level 1: food are pureed
Level 2: soft food that slay together as bolus avoiding spilling into airway
Level 3: chopped ground food items, such as, eggs and tender cooked vegetables.
(National Dysphagia Diet Task force)
Positioning :
Client should be positioned symmetrically with normal alignment b/w the head, neck,
trunk and pelvis.
Client is seated on a firm surface, like a chair.
Client’s feet are flat on the floor.
Client’s knees are at 90 degrees flexion.
Client’s trunk is flexzed slightly forward with back straight.
Equal weight bearing on both ischial tuberosities of hip.
35.
Compensatory techniques:
Chintuck (head flexion)
Head rotation (head turn)
Head tilt
Supraglottic swallow
Super-supraglottic swallow
Effortful swallow
Mendelsohn maneuver
Exercises:
Tongue hold
Shaker exercise
Strengthening of outer oral
36.
ENVIRONMENTAL MODIFICATION:
Adaptiveequipment and environmental modifications are often used as OT
interventions.
Home and workplace modifications will be necessary for most clients.
BATHROOM MODIFICATIONS:
Toileting- Rails and raised toilet seat/ toilet frame
Bathboard/ shower aids
Hoyer lift- preferably ceiling track
Long handled brush/ comb
Shaving, combing hair, cleaning teeth- weightand type of grip
Basin- wheelchair access
37.
FEEDING EQUIPMENT:
Elevateplate height, rocker knives to minimise the amount of active arm, wrist and
hand movement
Use plate guards/ high rimmed plates to prevent spillage and assist loading a fork
or spoon
Built-up utensils are helpful when grip strength declines.
Use only built-up spoons with handles
Try using straws instead of giving work to arm
38.
WHEEL CHAIR TRAINING:
Wheelchair prescription and mobility training in either a manual or powered
wheelchair are included in the OT intervention program.The wheelchair may require
a special seating system or supports to minimize the development of scoliosis, hip
and knee flexion contractures, and ankle plantar flexion deformity. Powered
wheelchairs are often recommended to conserve energy and decrease strain on
shoulders and trunk. Instruction in methods to maneuver power chairs can cause
overexertion, and this activity, like any other, must be graded for difficulty.
ARTICLE:
Danielle Hall, et, al. in 2020: Exploring the Benefits of Dynamic Arm Supports to
Enable Upper-Extremity Function in Men With Duchenne Muscular Dystrophy
(DMD): A Randomized Clinical Trial: This study is a longitudinal, randomized
clinical trial exploring the benefits of two wheelchair-mounted dynamic arm
supports (KINOVA O540 and JAECO WREX) in non-ambulatory individuals with
DMD experiencing upper-extremity weakness. Data collected will provide
information about upper-extremity movement patterns and performance, as
well as participant-reported outcomes regarding function, goal attainment,
independence, quality of life, and user satisfaction.
39.
FAMILY AND COMMUNITYREHAB:
A critical element in OT intervention is to help the client and family members find
meaningful activities in which to participate either as individuals or as a family.
For example, connecting with spiritual activities that engage clients and families on
the deepest level could be critical to a sense of hope and well-being.
Alternatively, encouraging families to use humor and to play and laugh together
can promote bonding, reduce fear and anxiety, and produce positive physical and
emotional feelings.
Client and family education is an important part of a team rehabilitation program.
A supportive approach to the client and family is helpful as function declines and
new mobility aids, assistive devices, and community resources become necessary.
40.
PSYCHOSOCIAL:
Psychosocial issuesrelated to MD involve the whole family. Parents go through
phases of shock, fear, and despair when the diagnosis is made and as the child
ages and function decreases (e.g., when the wheelchair is prescribed).
Encouraging parents not to be overprotective and to continue to promote their
child’s independence is an important aspect of therapy.
Further, therapists can anticipate times in the growing child’s life when
psychosocial support will be most essential and can offer education and support
during intervention sessions.
Some potentially disturbing developmental milestones for the client and family
occur when the child starts school (at approximately age 5), when the child loses
the capacity for independent ambulation (at 8 to 12 years), when adolescent social
life is limited, and, of course, when young adulthood arrives with the expectation
that death is imminent.
Referral to a psychologist, family counselor, or spiritual advisor during these times
should be considered
41.
CASE STUDY: Chronicpain in children, adolescents, and young adults (i.e., persons
between 8 and 20 years of age) with physical disabilities has not been widely
studied, but recent research indicates that chronic pain interferes with ADLs and
IADLs in young people with MD, cerebral palsy, spinal cord injury, acquired and
congenital limb deficiency, and spina bifida. Pain appears to be more prevalent in
female subjects than male subjects during the performance of daily routines, and it
interfered most often with physical activities, and it negatively affected mood.
McKearnan reported that the young people in this study frequently reported pain
associated with physical therapy, OT, and therapeutic home programs.They also
complained that the use of splints, orthotics, and prosthetics was sometimes painful.
Therapists must be aware of their client’s pain levels and modify activities, splints,
and orthotics as needed. Helping clients with MD to manage pain will most likely
lead to improved quality of life
(Pedretti’s Occupational therapy- practice skills for physical dysfunction- 7th
edition)
42.
SUMMARY
The motorunit consists of the lower motor neuron, neuromuscular junction, and
muscle. Some motor unit dysfunctions are reversible, and others are degenerative.
The role of the occupational therapist is to assess functional capabilities in all
occupational performance areas and contexts. ADLs and IADLs (including self-
care, home management, mobility, and work-related tasks), energy conservation,
work simplification, joint protection, spiritual approaches, and appropriate humor
may be used to restore or maintain function. Proper positioning, exercise
programs, and pain management techniques are used as indicated to facilitate
recovery and increase functional capacity. Orthoses, assistive devices,
communication aids, and mobility equipment and training in their use may be
necessary. Psychosocial considerations and client and family education are
important aspects of the OT program.
43.
FRAME OF REFERENCES
FRAMEOF REFERENCES:
Biomechanical FOR
Occupational FOR
Rehabilitation FOR
MOHO
44.
REFERENCES
Neurology andneurosurgery illustrated by Kenneth W. Lindsay.
Richard M Lovering et, al. in 2017: the muscular dystrophies: from
genes to therapies- NCBI.
Pedretti’s Occupational therapy- practice skills for physical
dysfunction- 7th edition













![CONGENITAL MUSCULAR DYSTROPHY
Congenital muscular dystrophies are a class of relatively rare conditions that present in
infancy. Because of the vagaries of the naming system, many forms of CMD are classified
with the limb-girdle muscular dystrophies (e.g., severe congenital autosomal recessive
muscular dystrophy [SCARMD]). The typical CMD cases are often those associated with
disturbances in the central nervous system. Newborns and infants with CMD have significant
weakness and up to a 10-fold increase in the blood level of the enzyme creatine kinase, a
general indicator of muscle damage. Clinical manifestations include muscle weakness,
hypotonia, delayed motor development, and severe contractures with consequent joint
deformities.](https://image.slidesharecdn.com/musculardystrophy-250613135456-71e6a054/75/Muscular-Dystrophy-pptx-and-management-of-OT-16-2048.jpg)