Downloaded 408 times




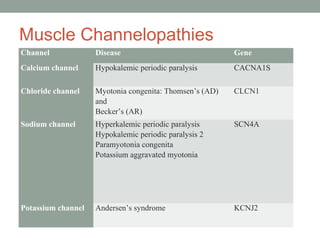









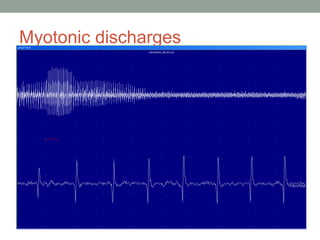
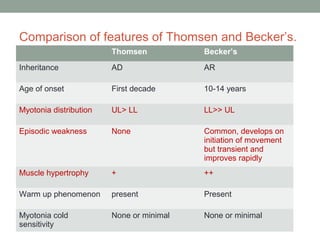





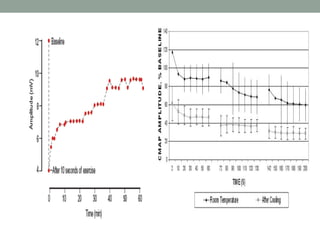





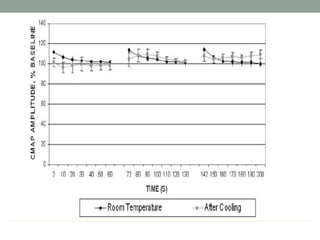
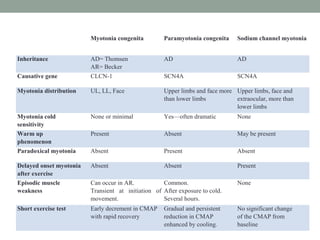



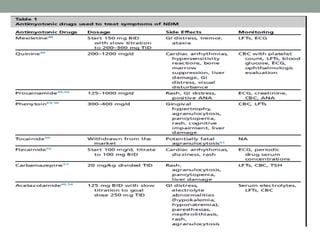







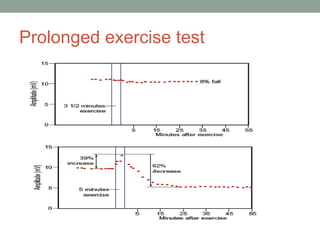

















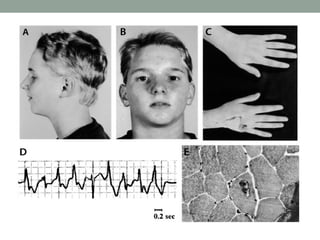
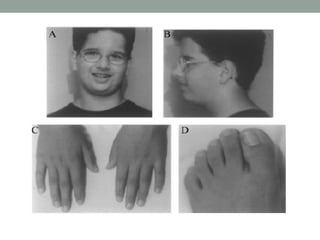



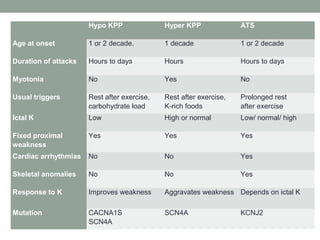












Muscle channelopathies are a group of rare inherited diseases caused by mutations in muscle ion channels. The document discusses several types of muscle channelopathies including non-dystrophic myotonias (myotonia congenita, paramyotonia congenita, sodium channel myotonia), and periodic paralyses (hypokalemic periodic paralysis, hyperkalemic periodic paralysis, thyrotoxic periodic paralysis). The key clinical features, genetics, pathophysiology, investigations, and management are described for each condition. Muscle channelopathies are characterized by episodic muscle stiffness, weakness, or paralysis triggered by factors like rest, exercise, cold, or meals and can be diagnosed via genetic testing and electrodiagnostic studies























































































