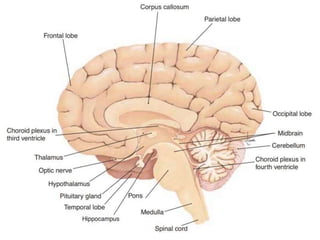
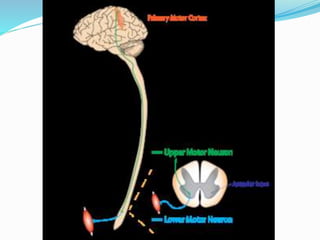
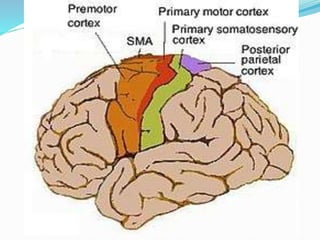
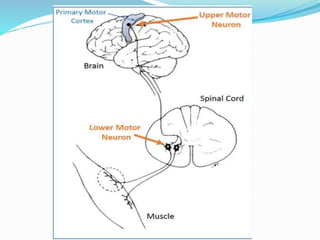
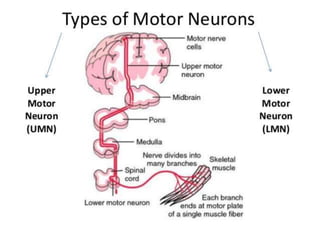
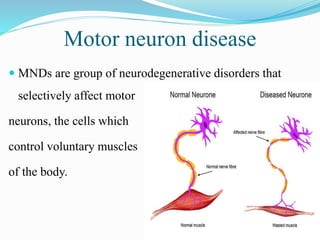
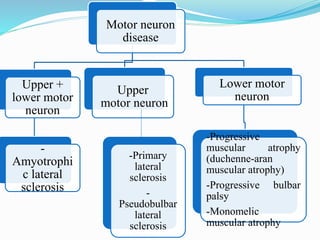
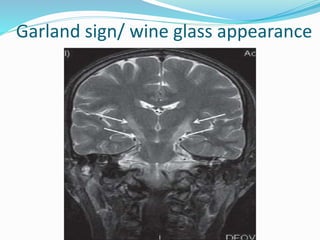
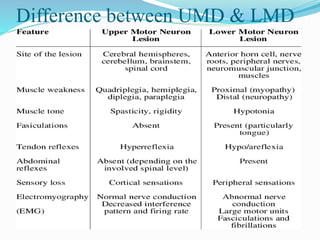
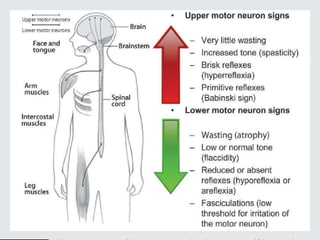
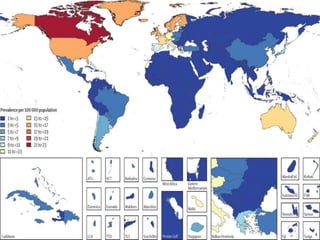
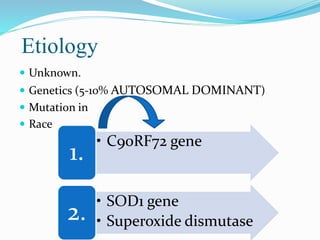
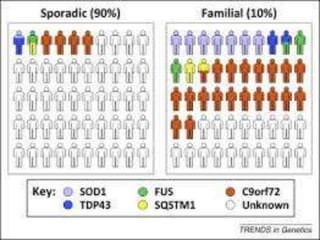
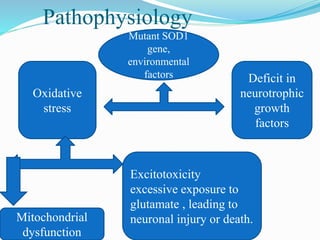
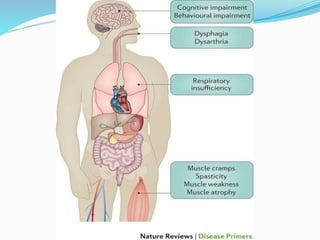
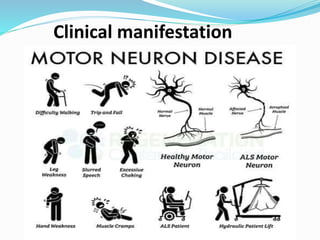
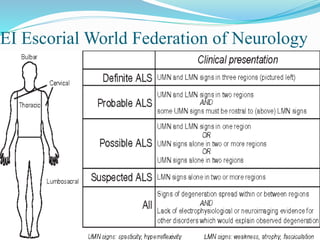
1. Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a progressive motor neuron disease that affects both upper and lower motor neurons.
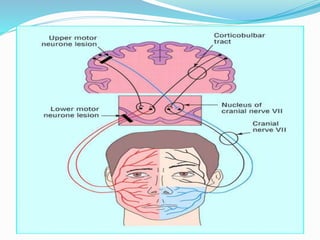
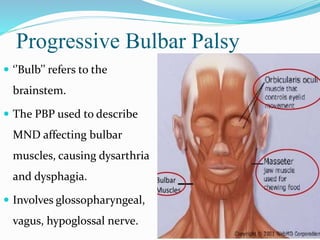
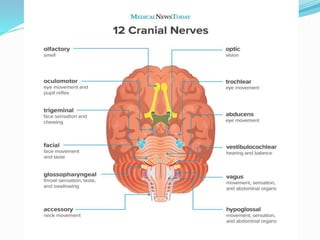
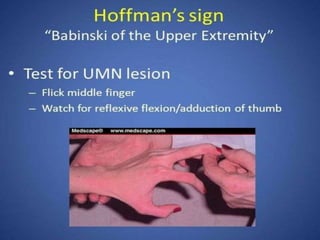
2. ALS is defined by evidence of both lower motor neuron degeneration causing weakness, wasting, and fasciculations and upper motor neuron involvement shown by spasticity and increased reflexes.
3. Riluzole is the only FDA-approved drug shown to modestly slow disease progression in ALS patients, extending survival by a few months. Edaravone was also approved in 2017 as an antioxidant therapy.






















































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)








