Downloaded 446 times
![Sickle cell anemia
Dr. Kalpana Malla
MD Pediatrics
Manipal Teaching Hospital
Download more documents and slide shows on The Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/hematology-sicklecellanemia-120108093300-phpapp02/75/Siickle-cell-anemia-1-2048.jpg)





































![Thank you
Download more documents and slide shows on The
Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/hematology-sicklecellanemia-120108093300-phpapp02/75/Siickle-cell-anemia-40-2048.jpg)

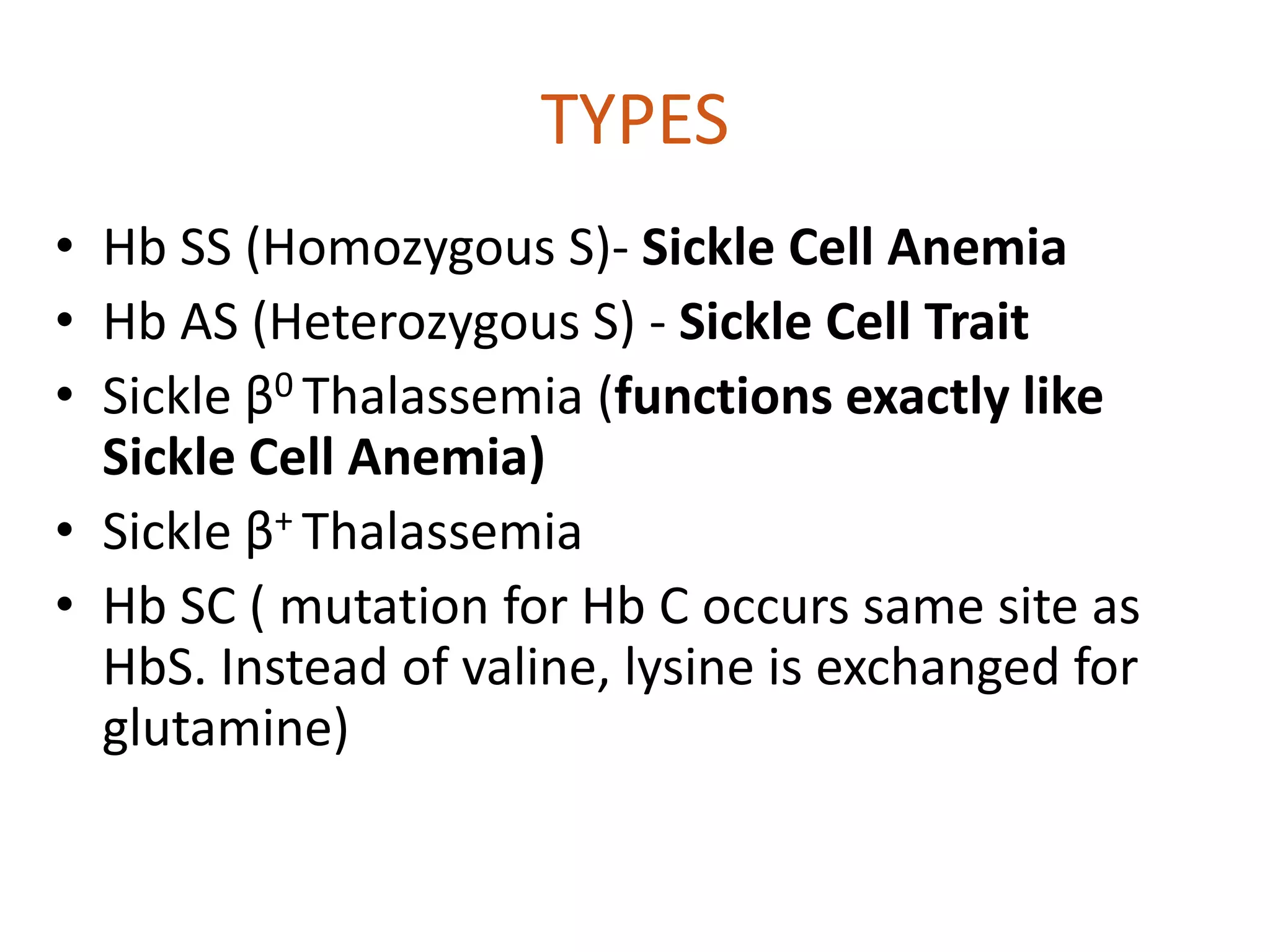
Sickle cell anemia is caused by a mutation in the beta globin gene that results in abnormal hemoglobin called HbS. People with two copies of the gene have sickle cell anemia and experience chronic anemia, pain crises, organ damage. Those with one normal and one mutated gene have the sickle cell trait and are usually asymptomatic but can experience complications in stressful situations. Management involves vaccination, antibiotics to prevent infection, pain medication, blood transfusions in acute situations, and hydroxyurea can reduce symptoms. Bone marrow transplant is the only cure but is high risk.











































































































