Downloaded 885 times
![Approach to a bleeding disorder,
Approach to thrombocytopenia,
Idiopathic thrombocytopenia (ITP)
Dr. Kalpana Malla
MD Pediatrics
Manipal Teaching Hospital
Download more documents and slide shows on The Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/hematology-bleedingdisorder-120108093006-phpapp01/85/Bleeding-disorder-1-320.jpg)







































































![Thank you
Download more documents and slide shows on The
Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/hematology-bleedingdisorder-120108093006-phpapp01/85/Bleeding-disorder-75-320.jpg)

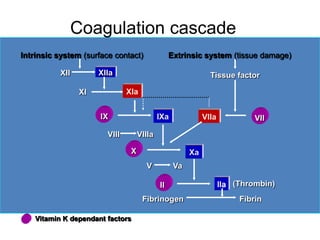
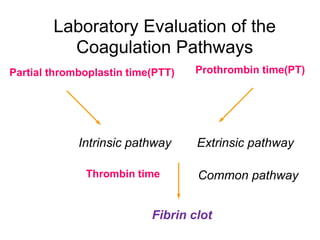
This document provides information on approaches to bleeding disorders and thrombocytopenia. It discusses idiopathic thrombocytopenic purpura (ITP), including that it is characterized by thrombocytopenia under 100,000/cm and shortened platelet survival due to the presence of antiplatelet antibodies. ITP most commonly affects children ages 2-10 years and presents with bruising of the skin and mucous membranes. Evaluation involves platelet counts and smears, while treatment options include corticosteroids, intravenous immunoglobulin, and anti-Rho(D) immune globulin.





































































































