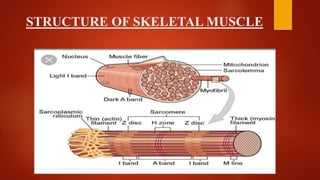
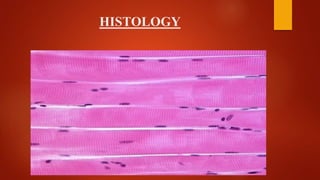
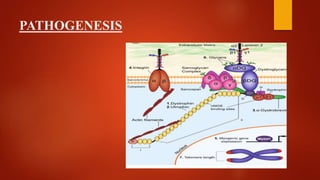
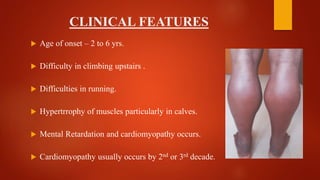
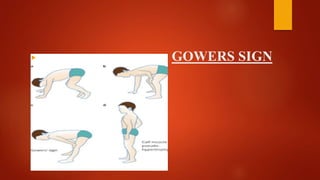
The document provides a comprehensive overview of myopathies, including their definitions, symptoms, classifications, and laboratory investigations. It details various types of muscular dystrophies, their clinical features, inheritance patterns, and diagnostic methods, along with treatment options for conditions like Duchenne and Becker muscular dystrophies. Additionally, it addresses associated disorders of muscle energy metabolism and mitochondrial myopathies.