Downloaded 303 times




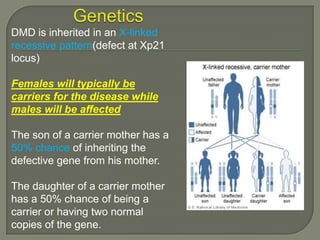
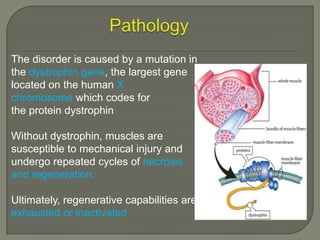
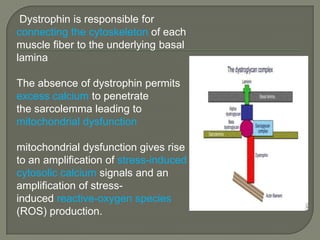
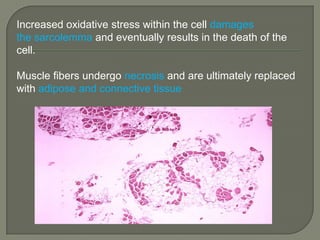
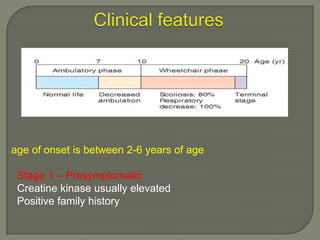

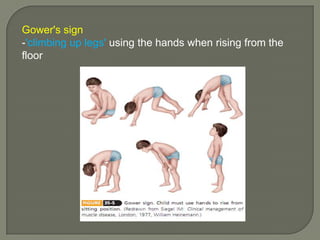


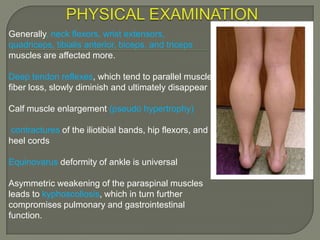
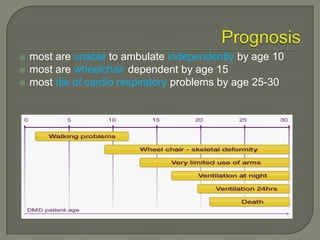
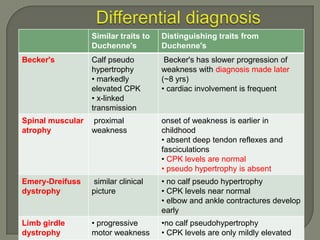


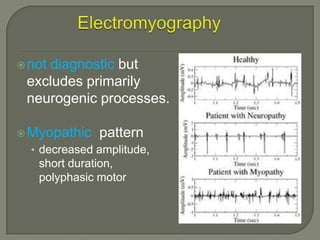

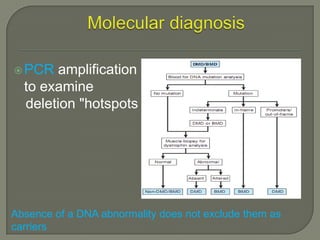



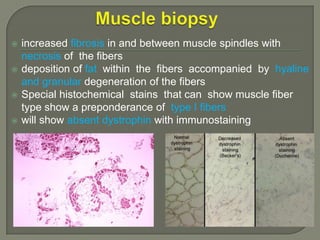
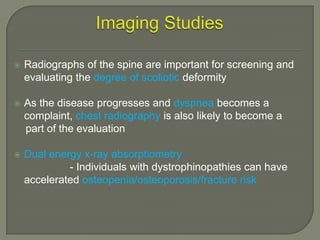




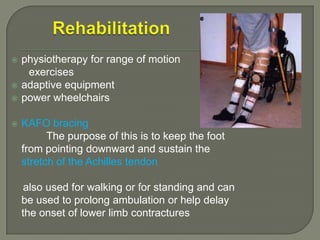



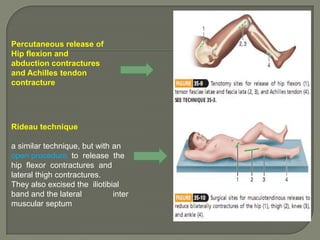

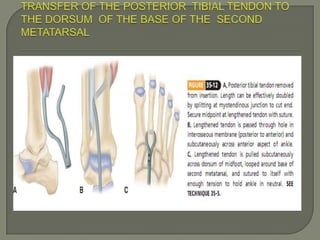


Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder characterized by progressive muscle degeneration and weakness. It is caused by the absence of the protein dystrophin due to mutations in the DMD gene. Symptoms usually begin between ages 2-6 and include difficulty walking, climbing stairs, and rising from the floor. Affected individuals lose the ability to walk by age 12 and often die in their 20s from respiratory or cardiac failure. Diagnosis involves testing for elevated creatine kinase levels and detecting dystrophin abnormalities through muscle biopsy or genetic testing. Currently there is no cure, but treatment focuses on symptom management through bracing, steroids, respiratory support, and physical therapy


















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)











