

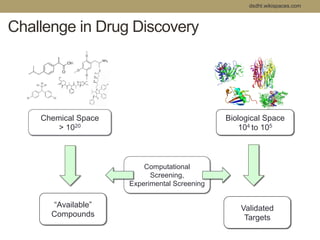

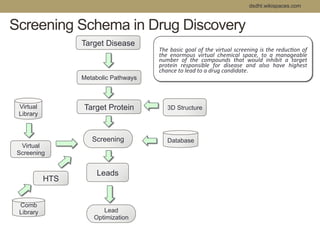
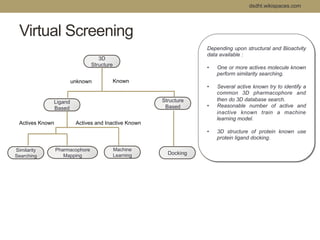
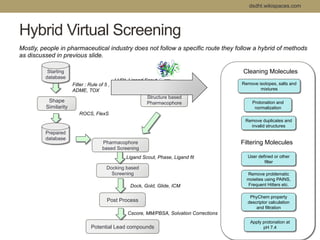




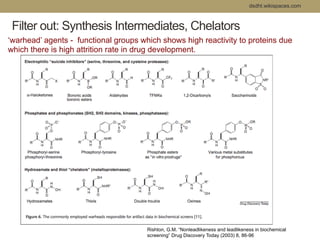
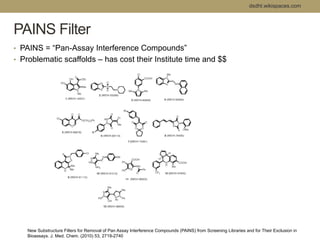
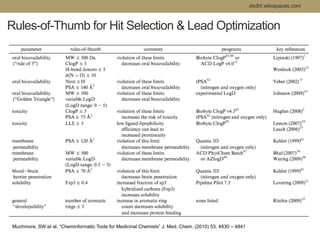
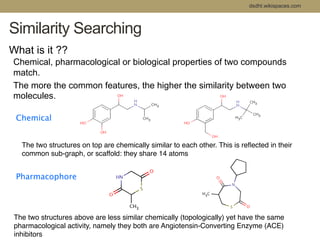
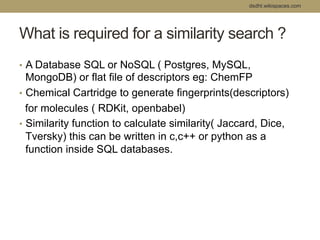



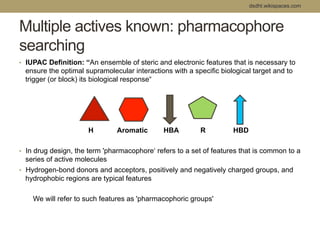
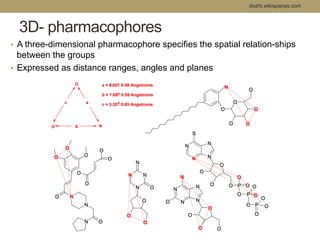
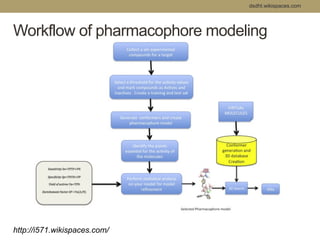


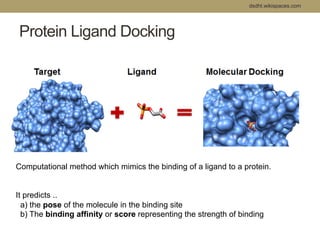


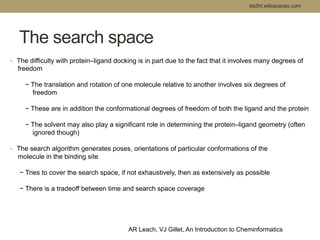


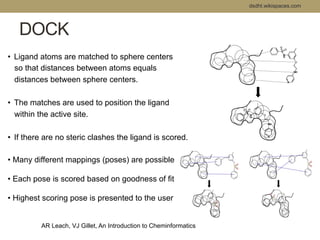





Virtual screening uses computer-based methods to filter large databases of chemical compounds to identify a subset of compounds that are most likely to bind to and activate a target linked to a disease. It helps address the challenge of exploring the vast chemical space compared to the limited number of compounds that can be experimentally screened. The document discusses various virtual screening methods including ligand-based approaches like similarity searching and pharmacophore modeling as well as structure-based approaches like molecular docking that predict binding orientations. It also covers best practices for applying filters to select for drug-like and lead-like compounds.











































![(Form r) sse (ae os) [howpk.com]](https://cdn.slidesharecdn.com/ss_thumbnails/formrsseaeoshowpk-160124072004-thumbnail.jpg?width=640&height=640&fit=bounds)































































