Downloaded 2,146 times









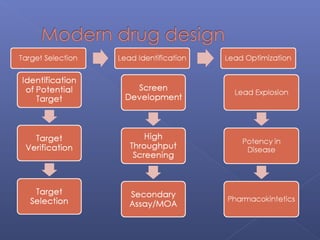






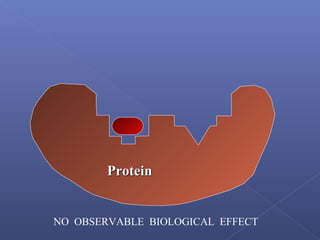
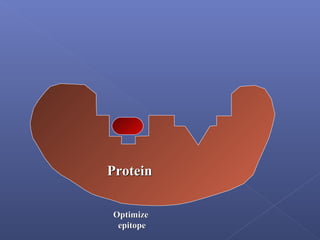
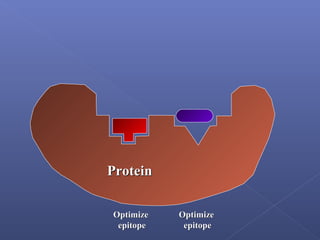
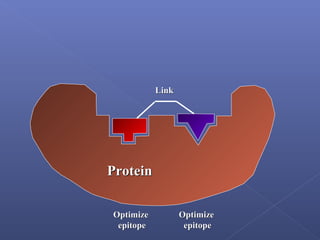


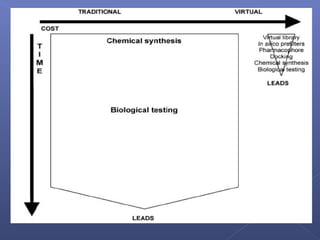


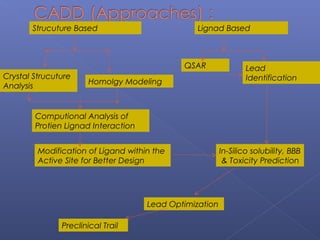
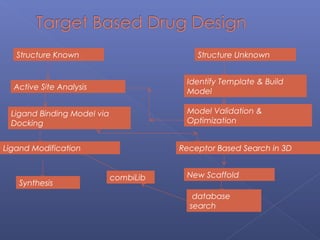
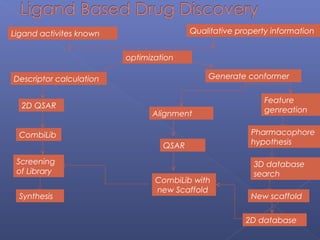



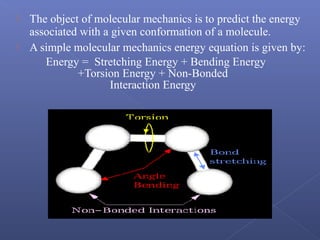
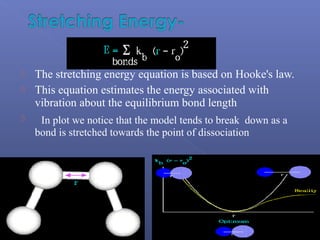
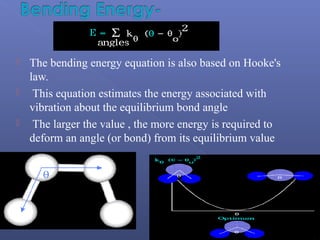

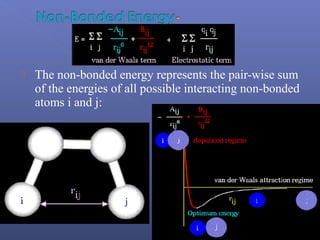




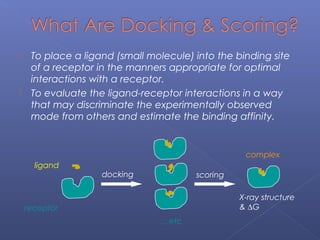





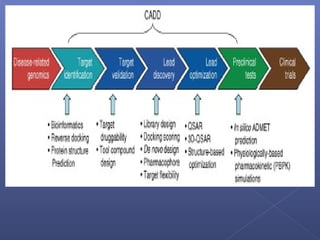
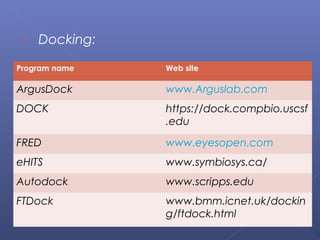

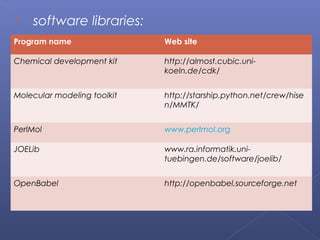




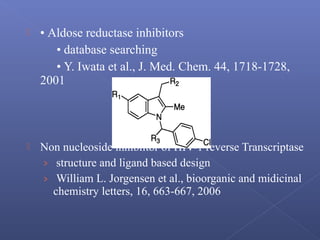







This document provides an overview of the history and methods of drug discovery, including traditional and computer-aided approaches. It discusses the traditional drug discovery life cycle from hit identification through random screening and the use of natural products and synthetic chemicals. It then introduces computer-aided drug design (CADD) and describes how it can be used throughout the drug discovery process, including structure-based design, ligand-based design, and de novo design to speed up screening and enable more rational drug design. It also lists some advantages of CADD over traditional methods and examples of drugs successfully developed using these approaches.

























![OLIGONUCLEOTIDE THERAPY [ TECHNIQUES, APPLICATIONS]](https://cdn.slidesharecdn.com/ss_thumbnails/66-191220063512-thumbnail.jpg?width=640&height=640&fit=bounds)
















































![Getting Started with Apache Spark: Big Data Made Simple [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/apachesparkgettingstarted-260203175547-8361bcc3-thumbnail.jpg?width=640&height=640&fit=bounds)









