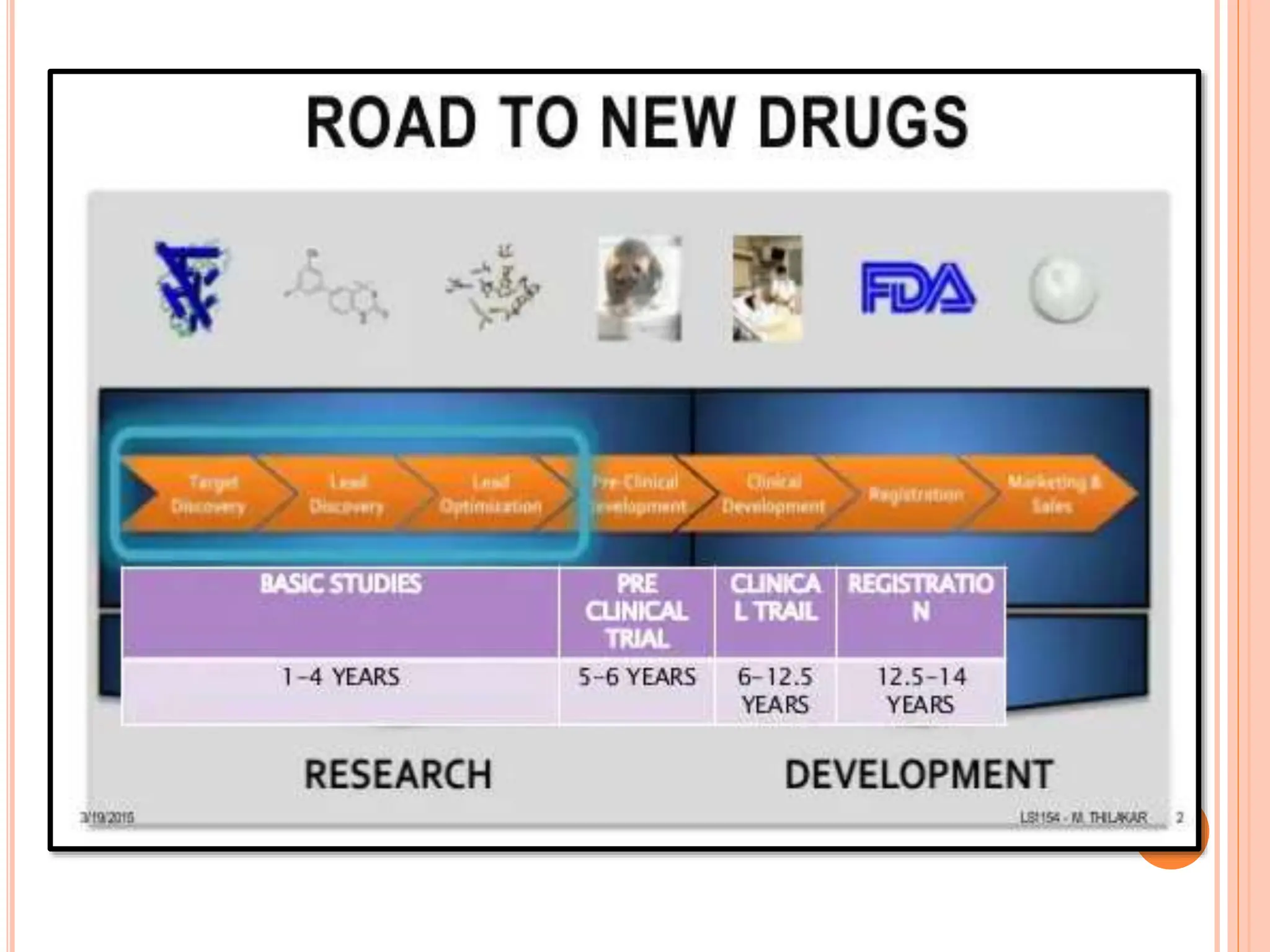
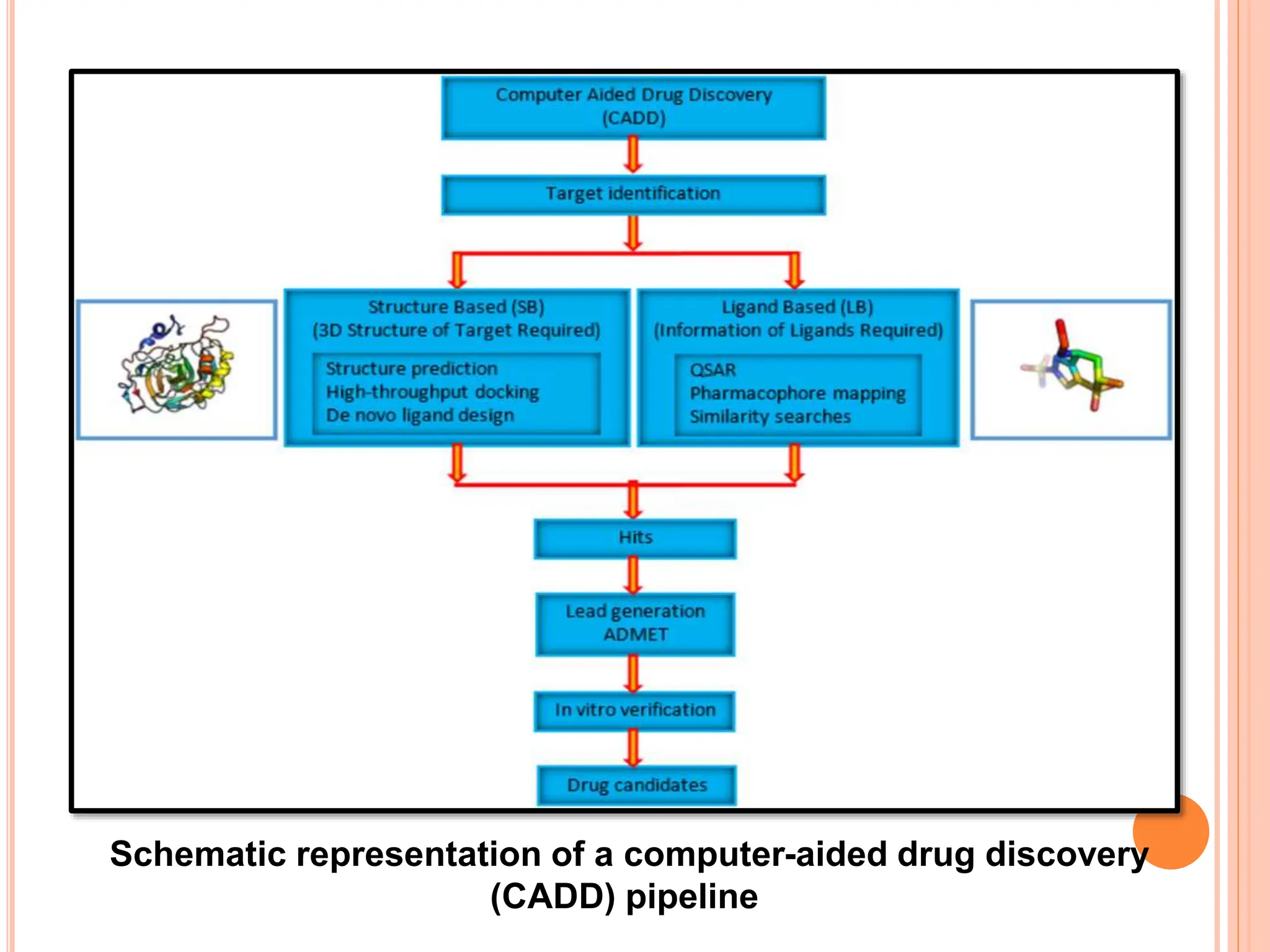
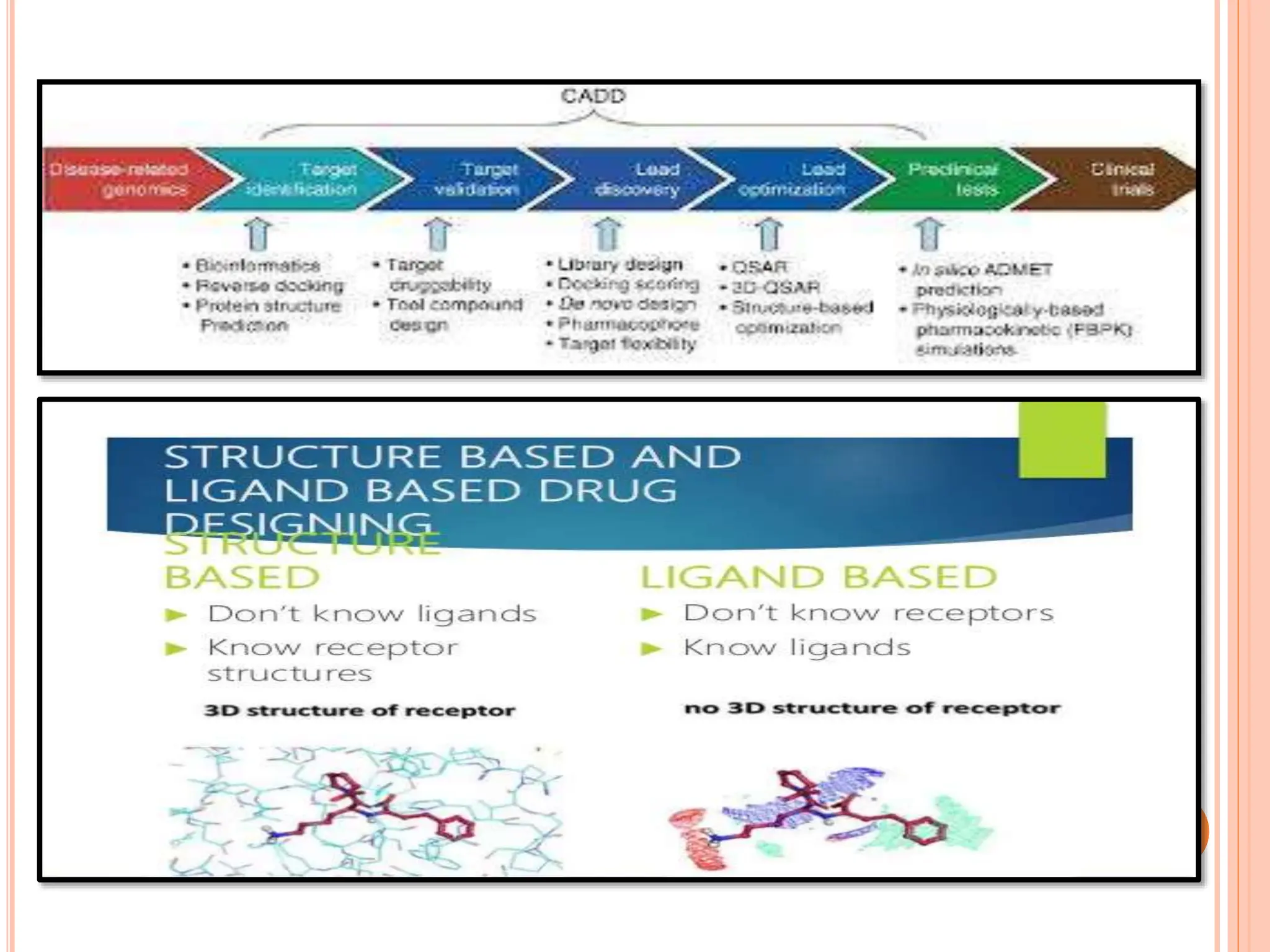
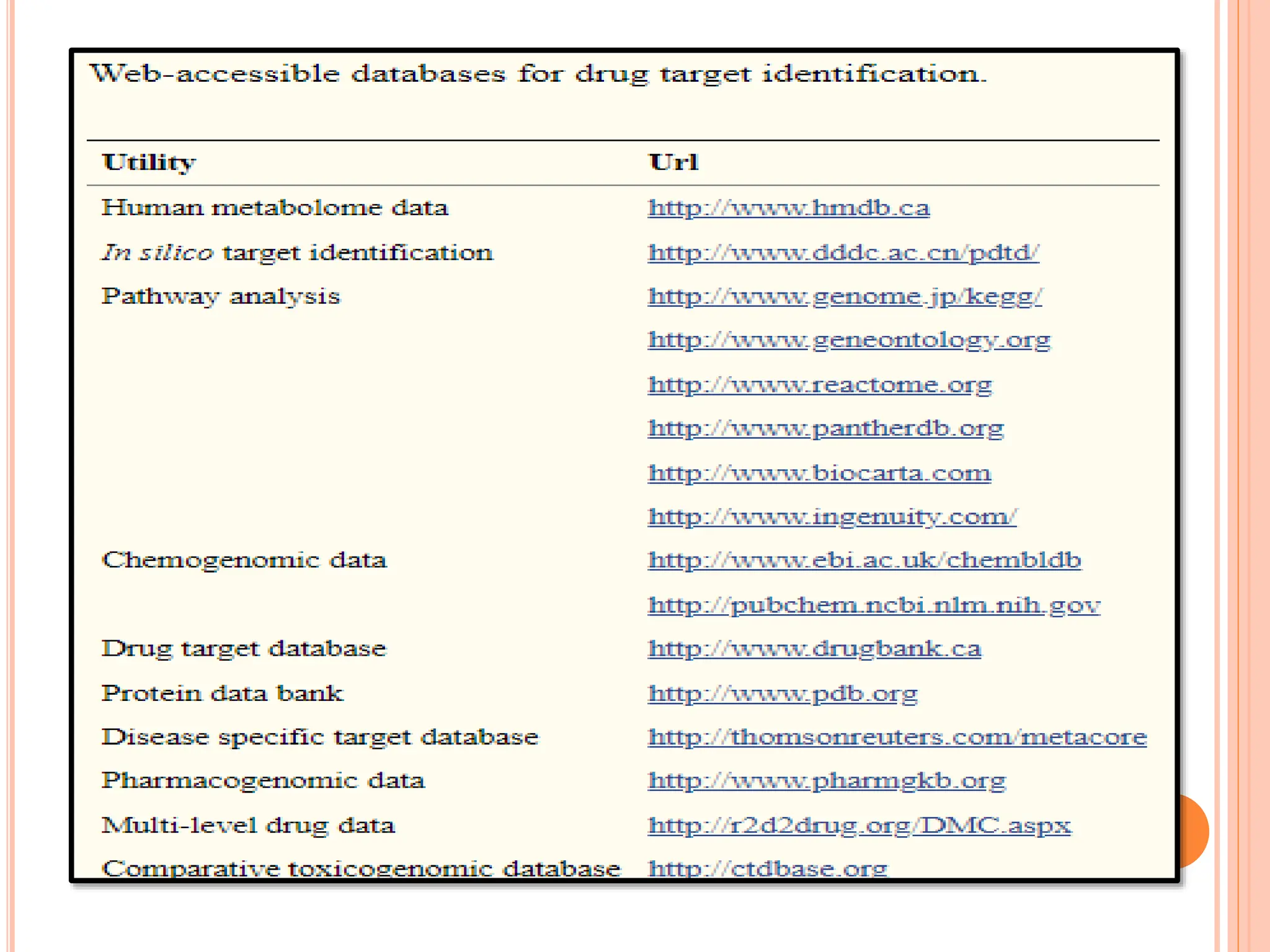
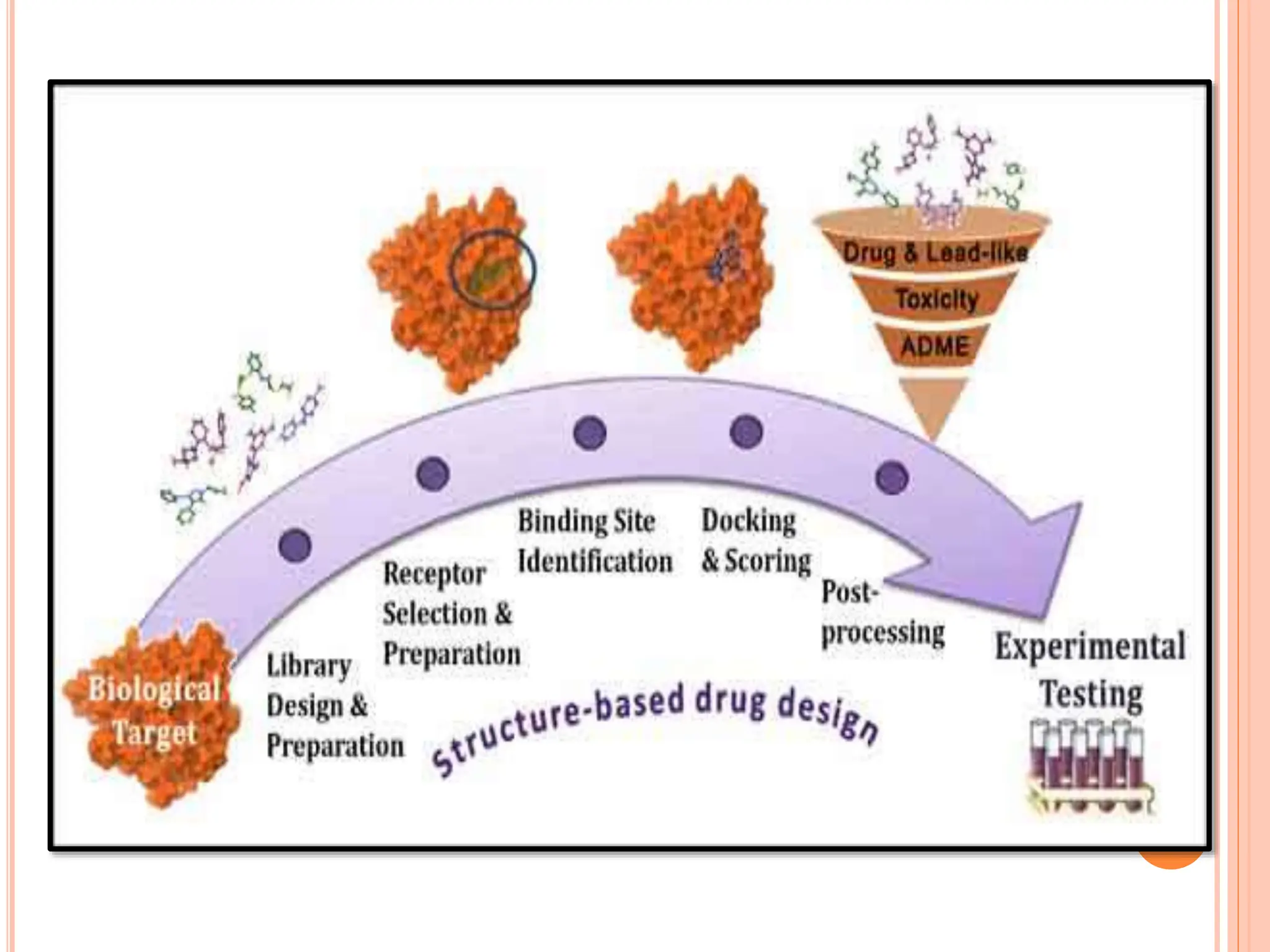
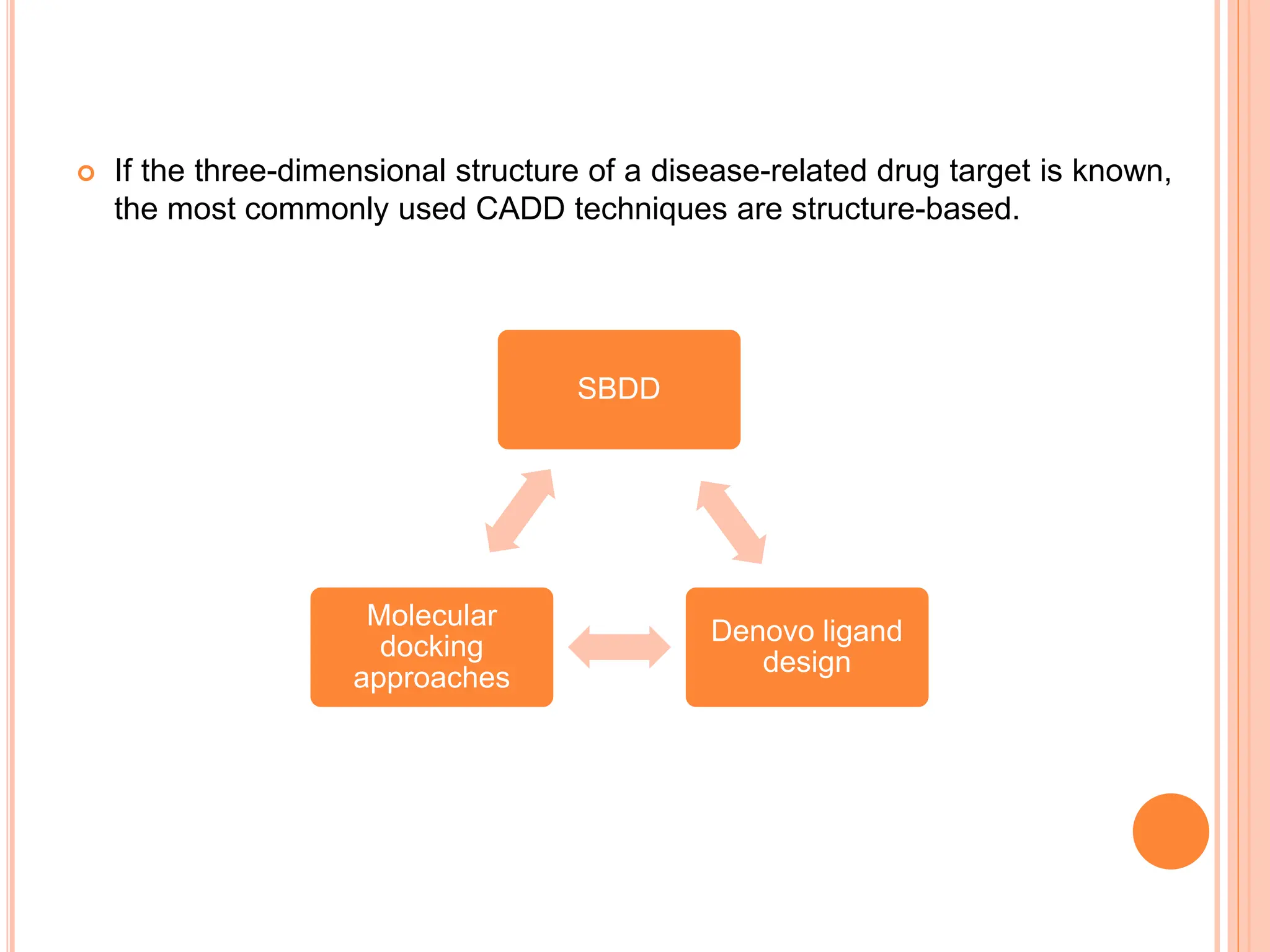
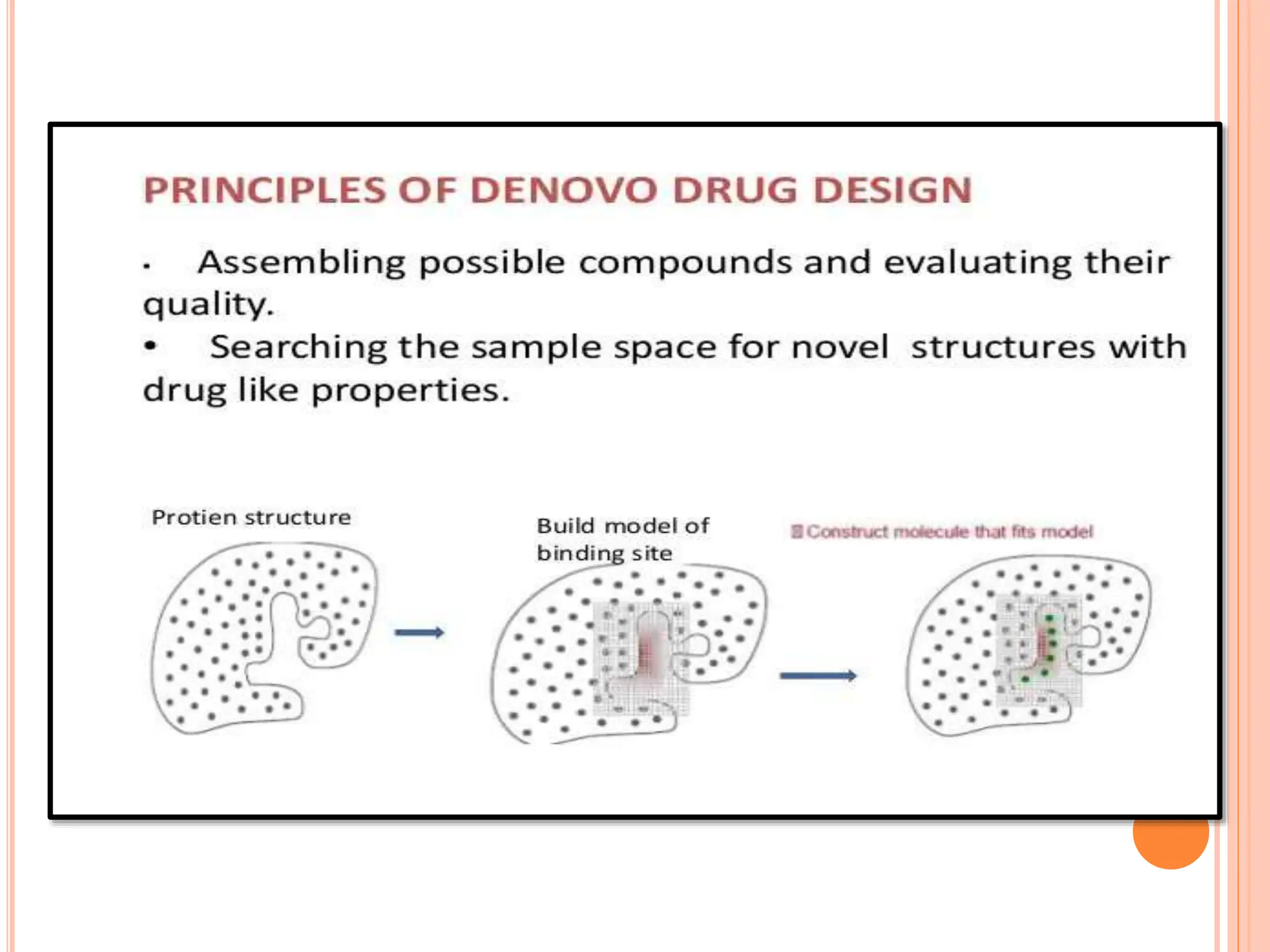
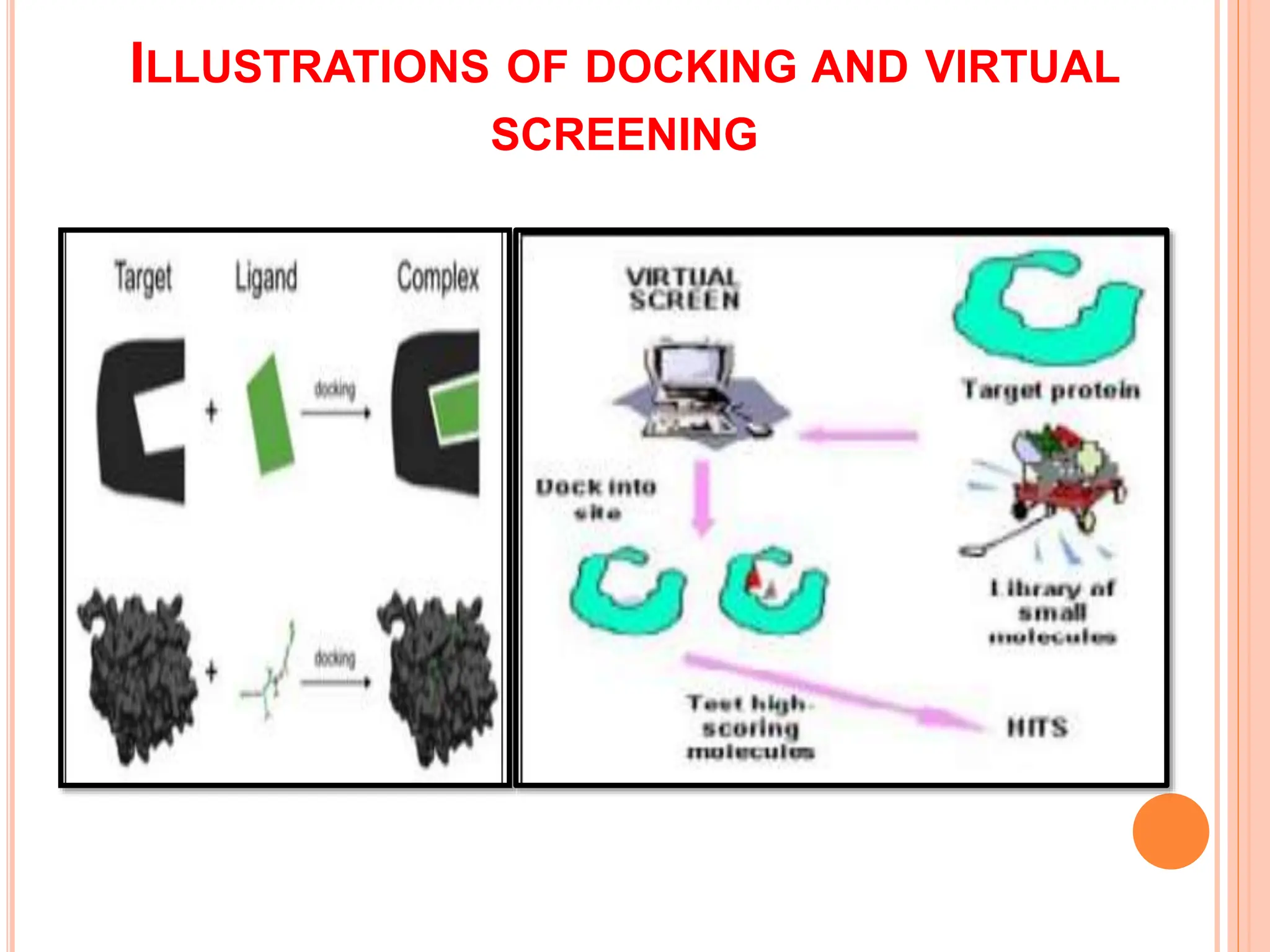
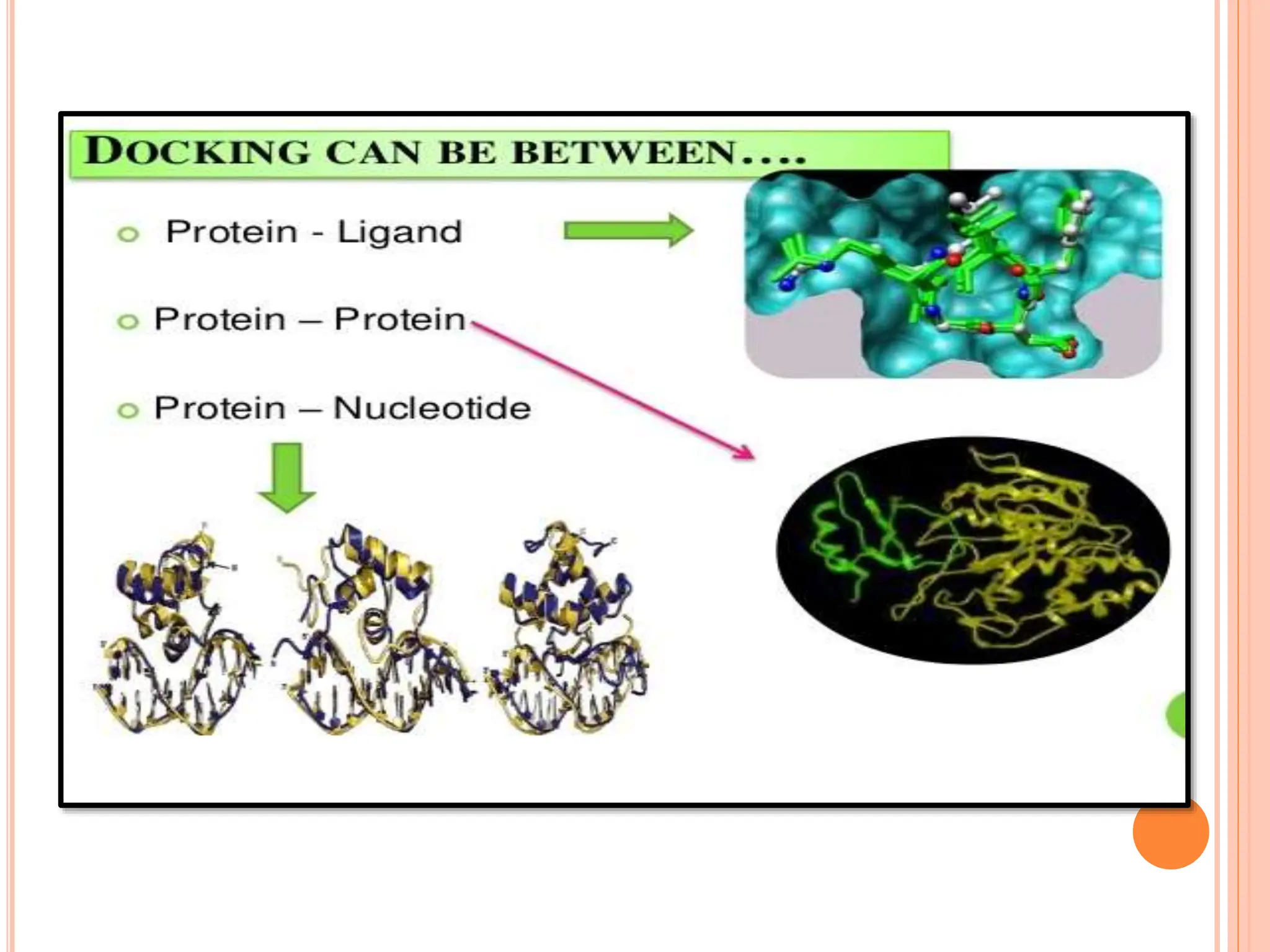
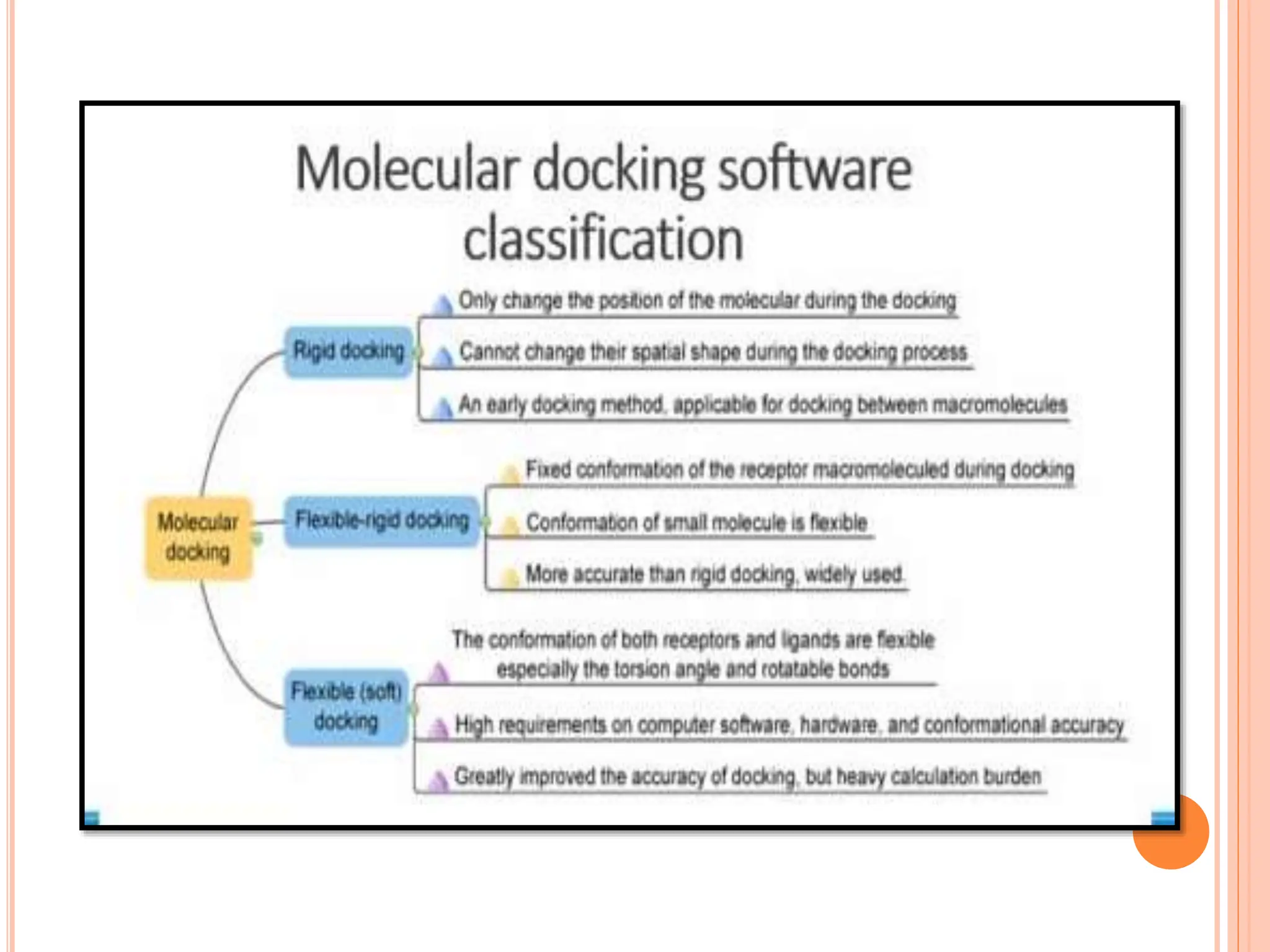
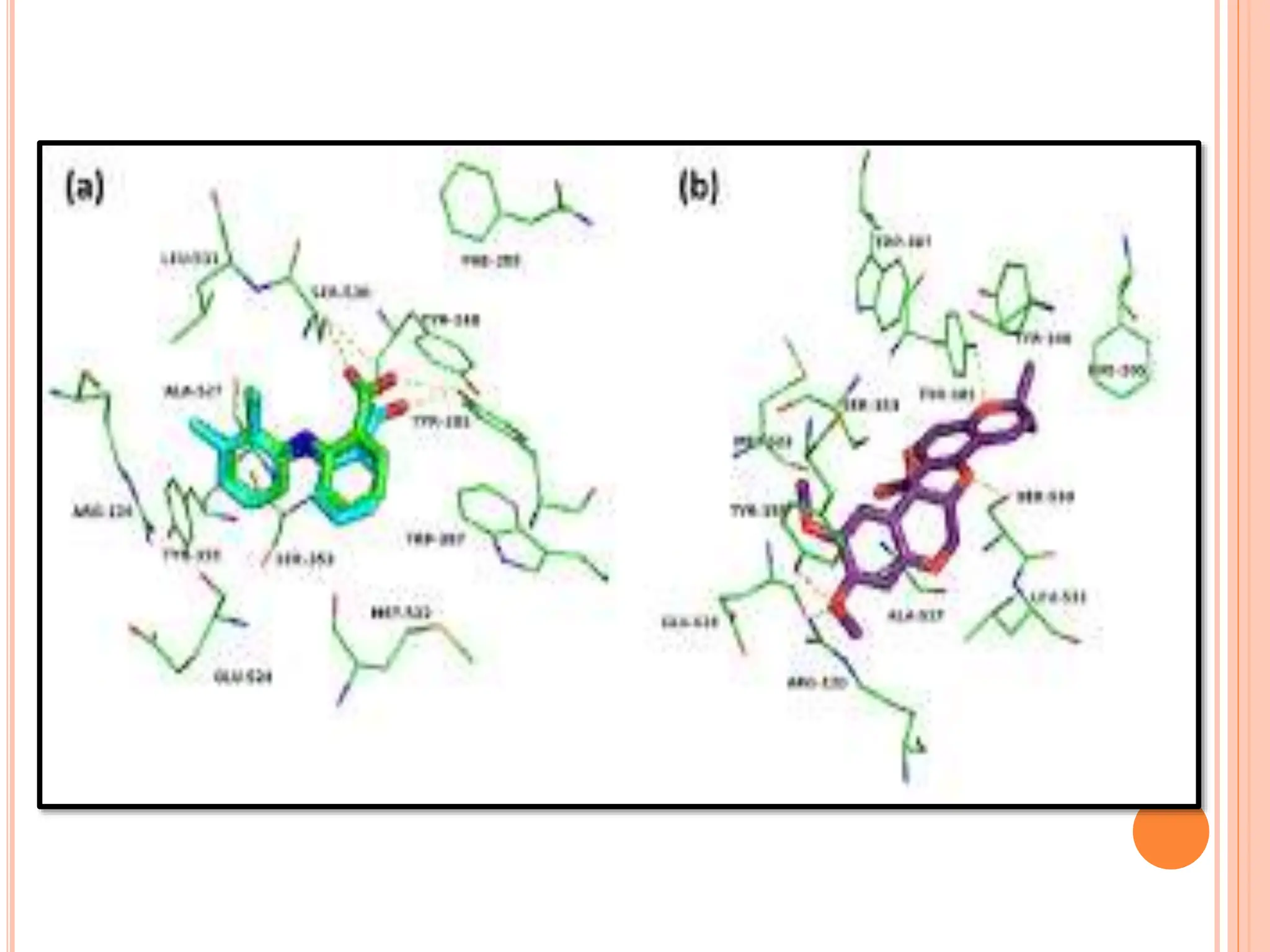
The document discusses the significant role of computational tools in drug discovery, highlighting that the drug development process is lengthy, costly, and complex, often requiring effective target identification and validation. It describes various computational methods such as computer-aided drug design (CADD), both structure-based and ligand-based approaches, and emphasizes the importance of binding pocket identification, molecular docking, and lead optimization in the drug development pipeline. Overall, the advancements in computational methods are essential for enhancing drug discovery and addressing unmet clinical needs.