














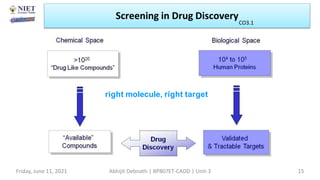







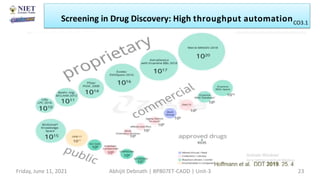





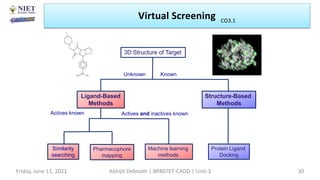
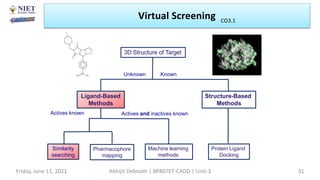
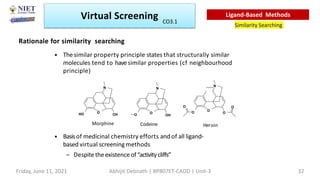

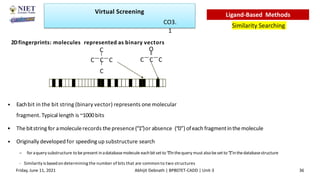
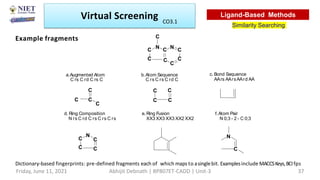
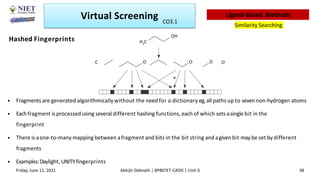
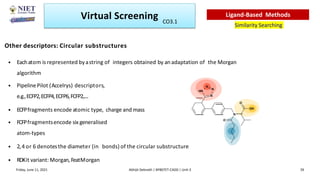

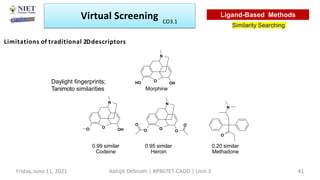
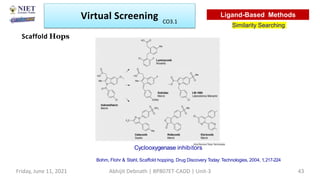
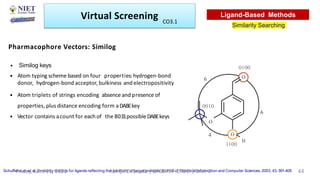
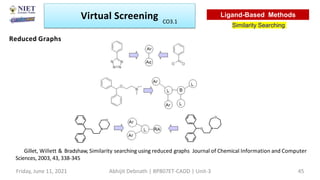

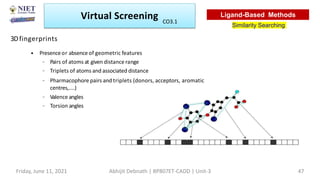







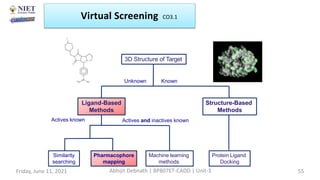

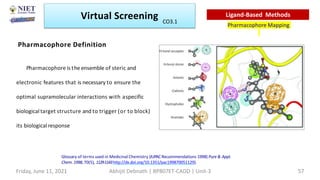
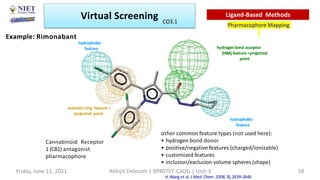
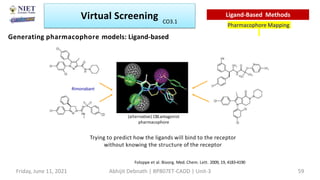



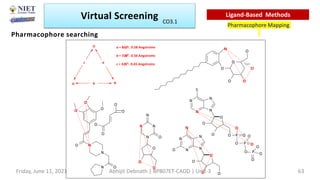
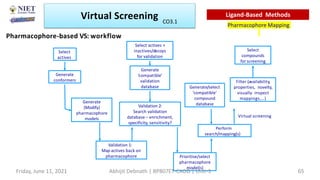


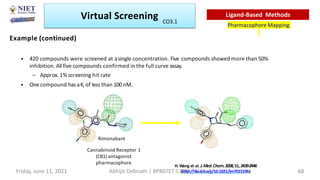
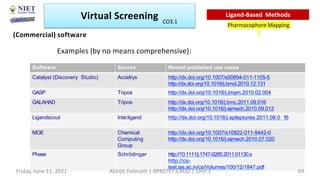
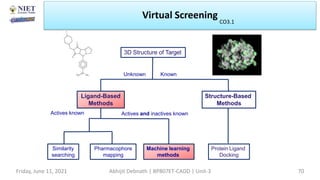
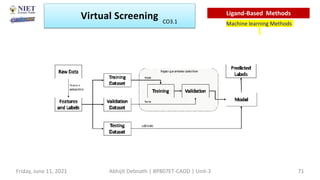

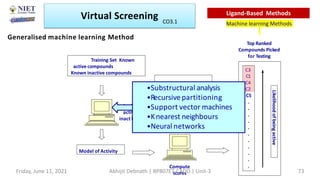


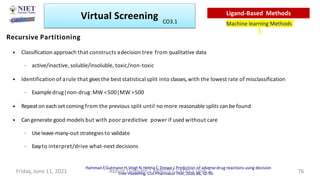
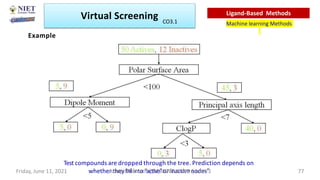
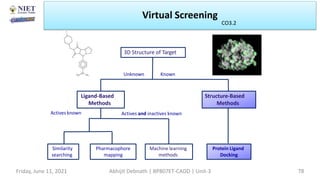


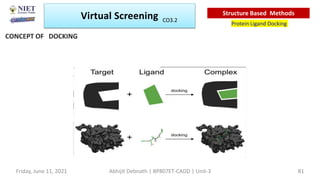
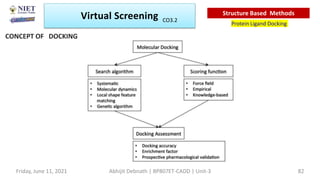


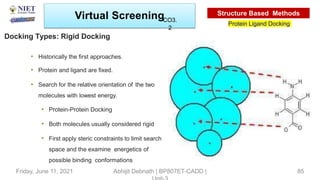
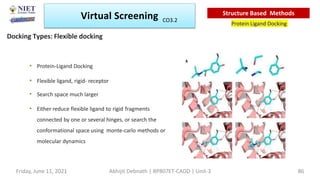












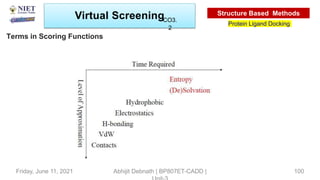






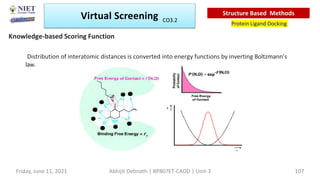
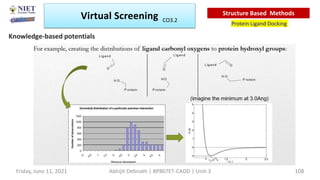

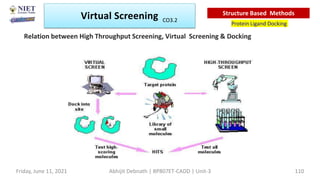

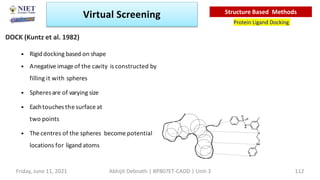
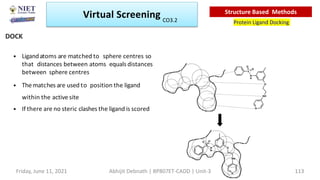
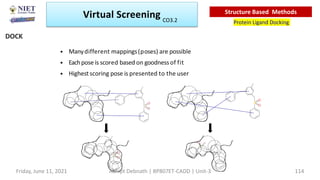



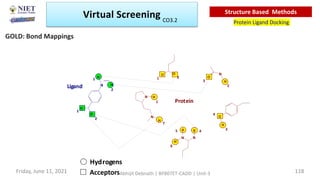

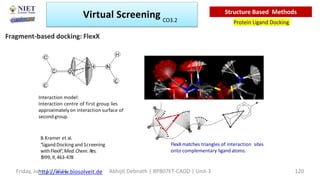
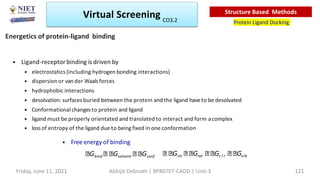




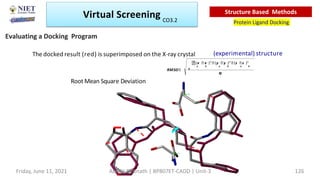
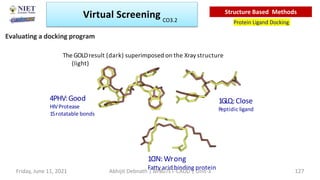



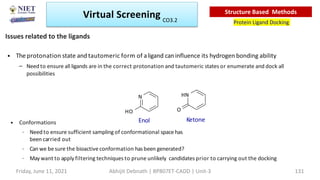

















The document discusses molecular modeling and virtual screening techniques, focusing on drug likeness screening, pharmacophore mapping, molecular docking, and de novo drug design. It outlines course objectives, expected outcomes, and prerequisites for pharmacy students, emphasizing the importance of virtual screening and high-throughput screening in drug discovery. Additionally, it covers key concepts like drug likeness, the rules for evaluating drug-like properties, and various methods for similarity searching in compound databases.























































































