Downloaded 505 times



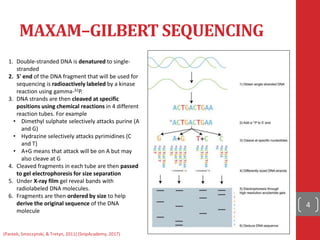

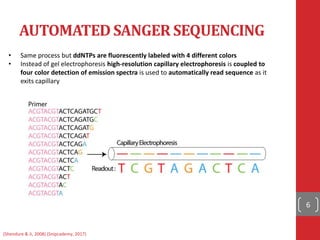

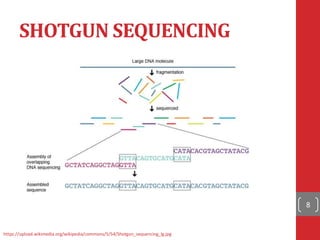


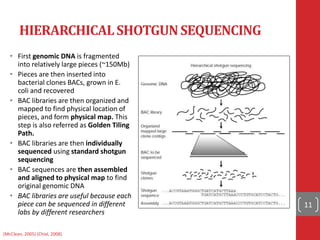









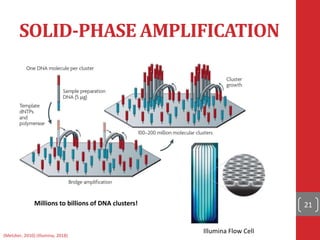





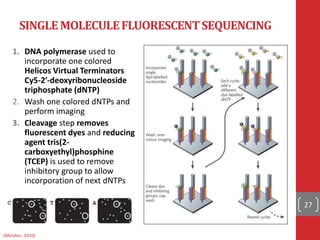
















The document provides a comprehensive overview of DNA sequencing, detailing the evolution from first-generation methods like Maxam-Gilbert and Sanger sequencing to advanced next-generation sequencing (NGS) technologies. It outlines various sequencing techniques, principles, and methods involved in library preparation, amplification, and sequence alignment. Additionally, it compares different NGS platforms and their specifications, highlighting their capabilities and advancements in genomic sequencing.



































































