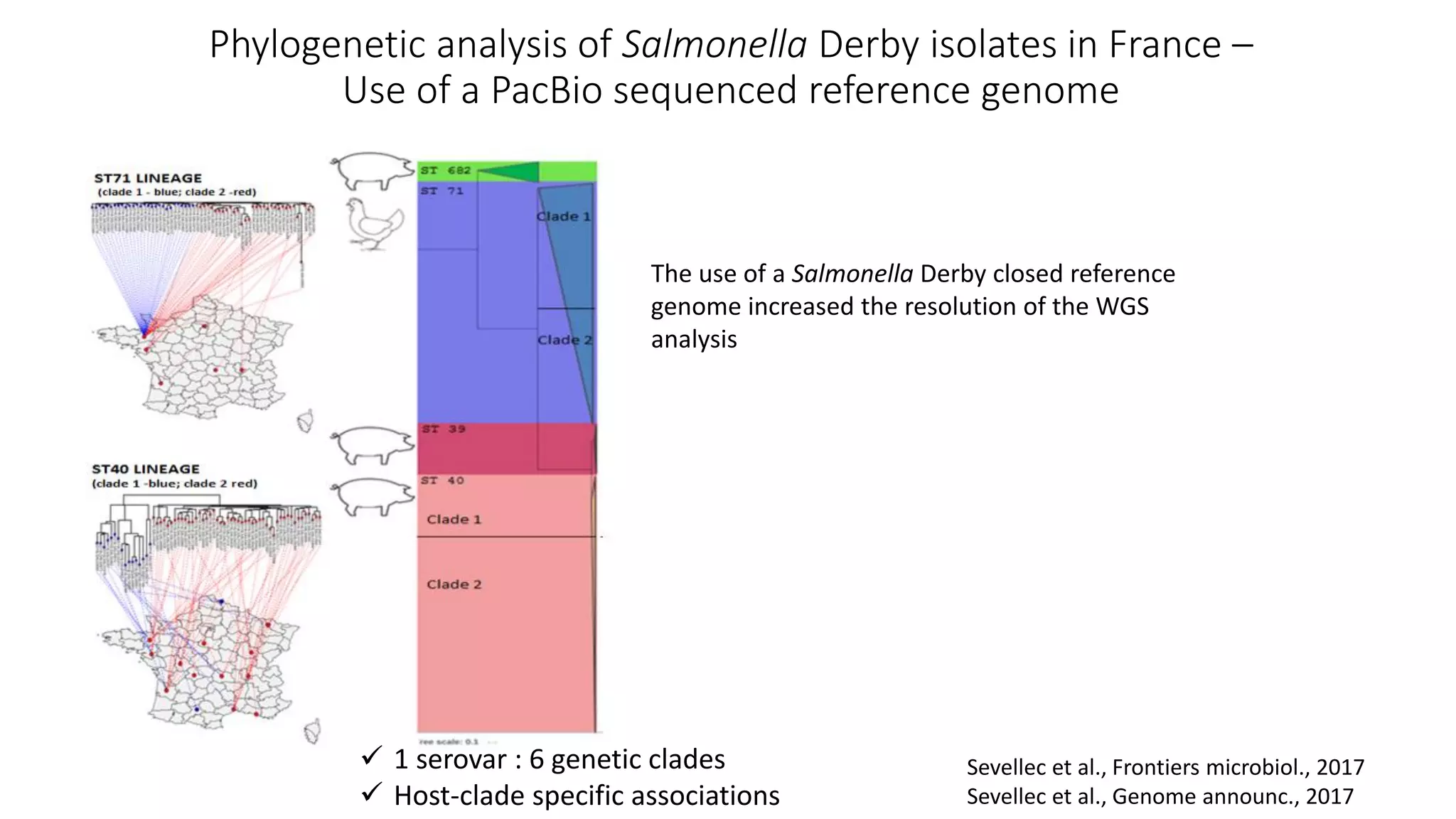
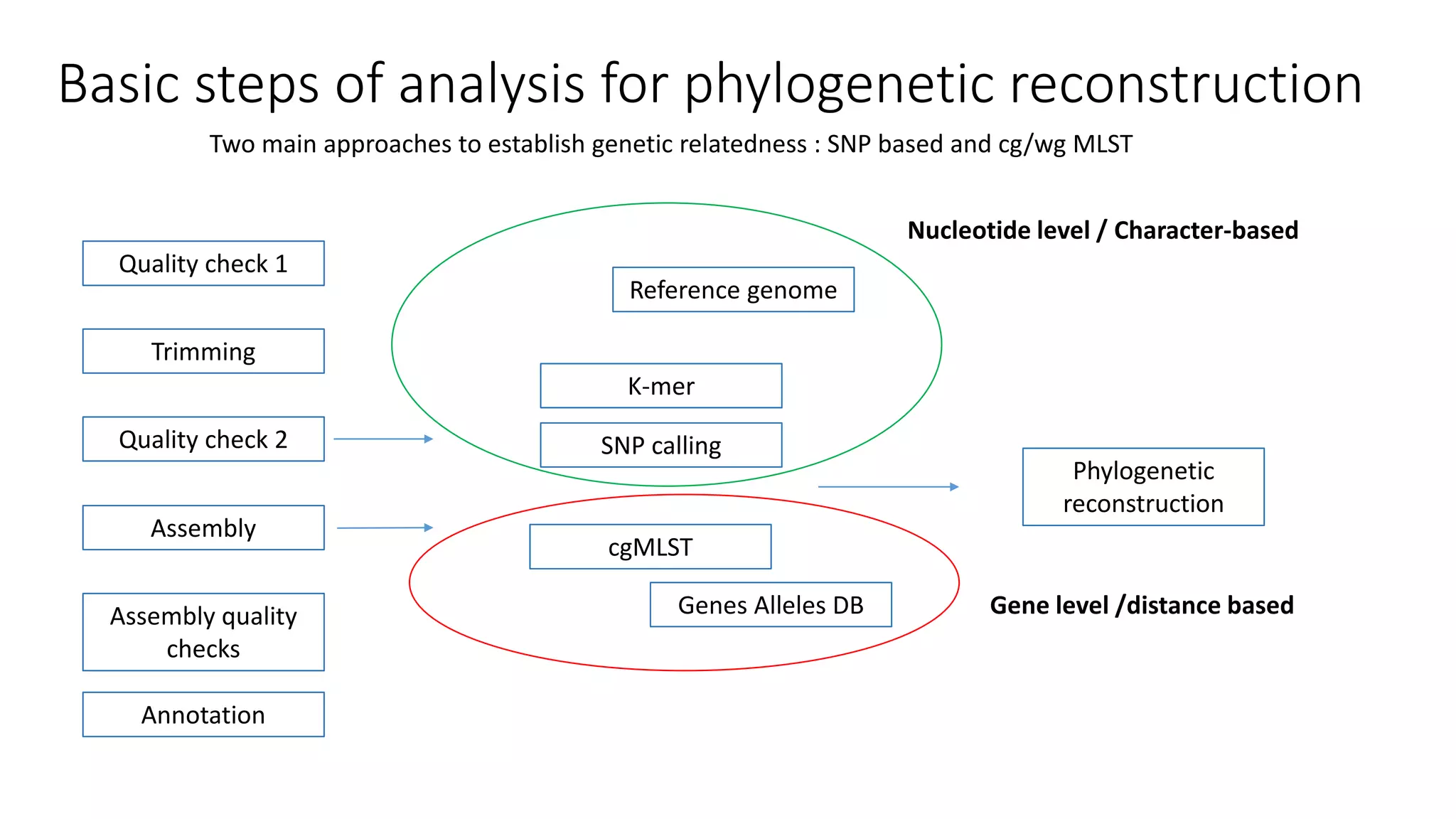
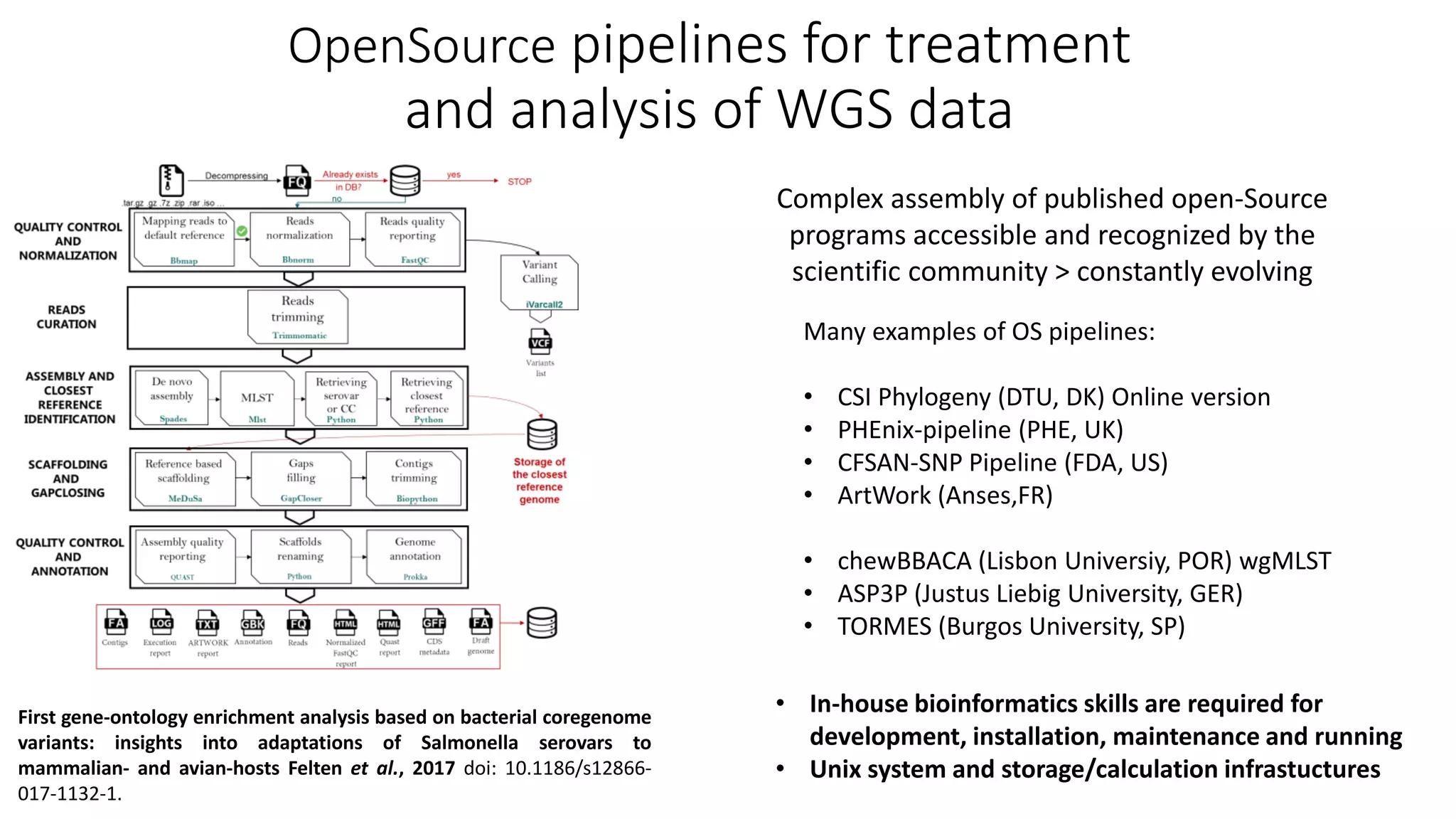
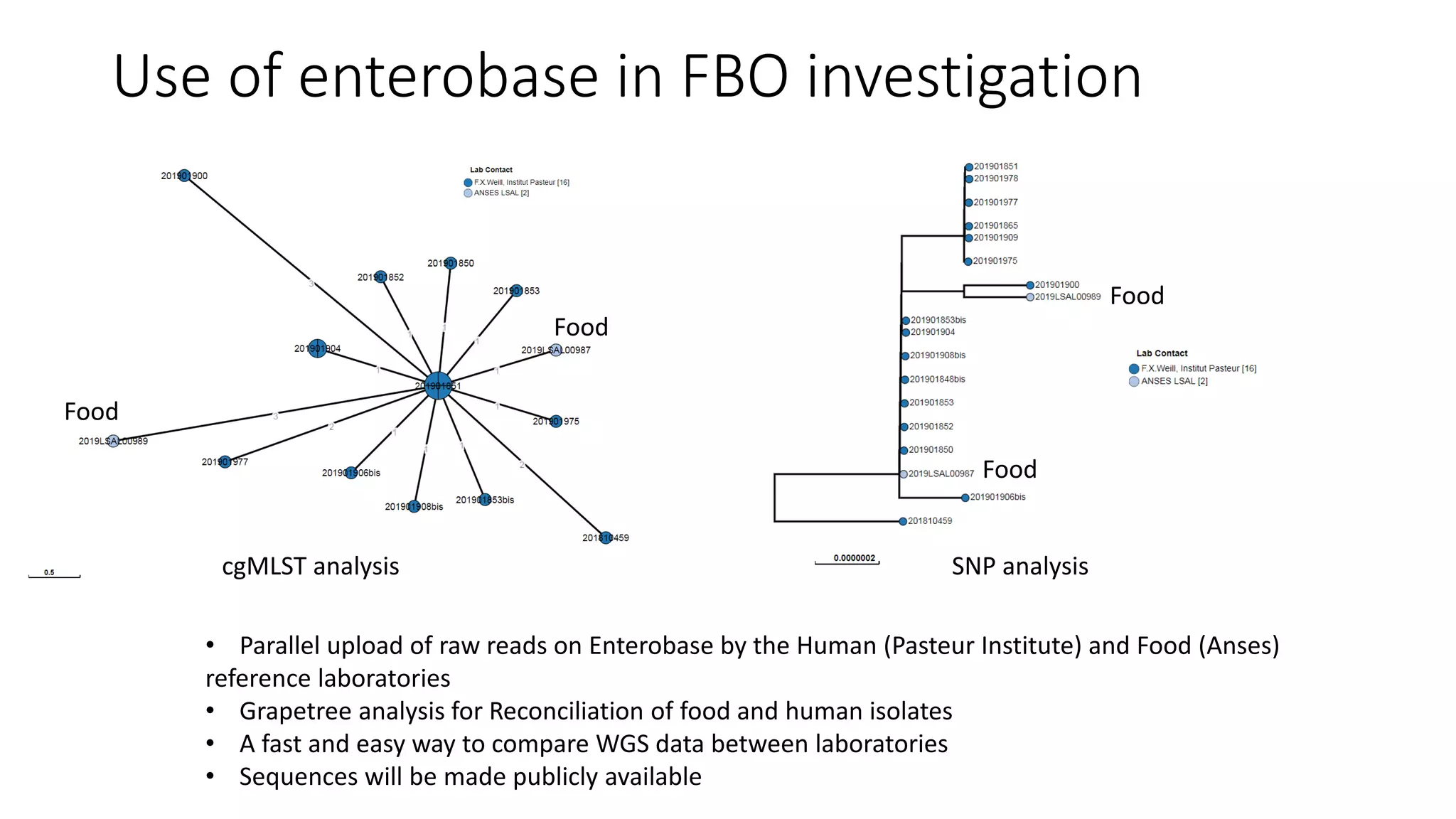
Download as PDF, PPTX










![• Three SNP-calling pipelines of major institutes (DTU, PHE,
FDA) tested on the same dataset
• Global epidemiological concordance, however
• Highly different SNP distances returned : 2-4 / 3-6 / 12-24
Benchmarking of SNP calling pipelines
Comparison of SNP-based subtyping workflows for bacterial isolates using WGS data, applied to Salmonella enterica serotype Typhimurium and serotype
1,4,[5],12:i:. Saltykova et al., 2018. doi: 10.1371/journal.pone.0192504
CSI
(DTU, DK)
PHEnix
(PHE, UK)
CFSAN
(FDA, US) > Results are sensitive to data quality, collection of isolates,
pipelines, parameterization … Universal SNP distance cutoff
values to decide about a link between isolates cannot be
determined](https://image.slidesharecdn.com/2-mistouoecd24thjune2019-200702151143/75/Overview-of-the-commonly-used-sequencing-platforms-bioinformatic-search-tools-and-databases-Michel-Yves-Mistou-14-2048.jpg)








The document discusses advancements in sequencing technologies and bioinformatics utilized in microbial genomics, highlighting platforms such as Illumina and BGI's innovations. It emphasizes the significance of short-read technologies for high-throughput applications while acknowledging the limitations of long-read technologies like PacBio and Oxford Nanopore, particularly their suitability for routine microbial genomics. Additionally, it covers the regulatory context and the need for standardized protocols and databases to ensure comparability and harmonization in genomic analysis for epidemiological purposes.












































































