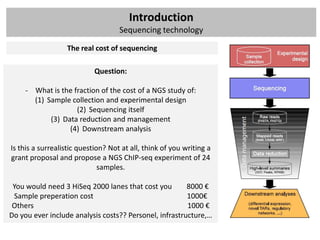
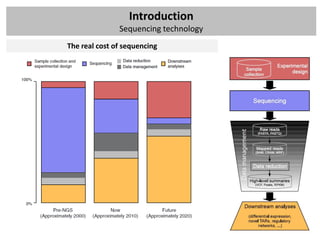
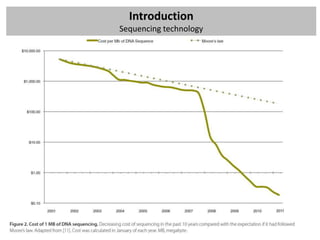
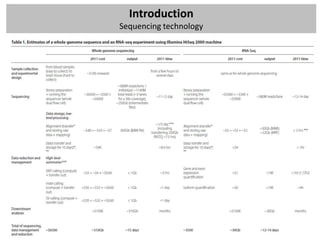
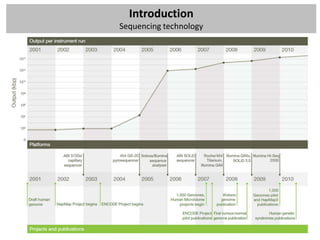
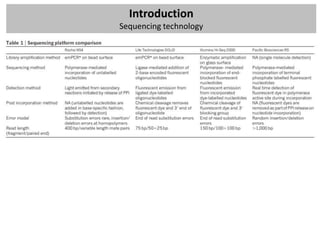
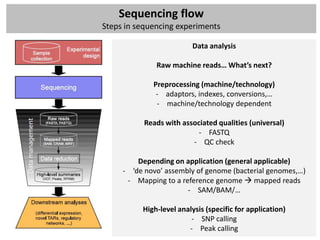
The workshop covers the main principles and data formats in sequencing data analysis, detailing steps from sample preparation to high-level analysis including various file types like fastq, bam, and vcf. It emphasizes cost considerations in next-generation sequencing (NGS) studies and provides practical exercises involving data retrieval and manipulation using command line tools. The document also discusses mapping techniques and common data formats used in genomic analysis, highlighting their applications in downstream analysis and visualization.












![Sequencing data formats
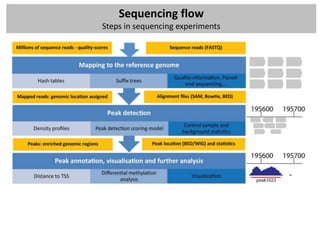
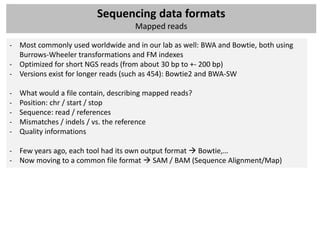
Mapped reads
- Mapping: ‘align’ these raw reads to a reference genome
- Single-end or paired-end data?
- How would you align a short read to the reference?
- Old-school: Smith-Watherman, BLAST, BLAT,…
- Now: mapping tools for short reads that use intelligent indexing and allow mismatches
Algorithm
Other features
Hash table Suffix tree Merge sorting
Hash Hash Enhanced
Program Reference Suffix tree FM-index Merge sorting Colorspace 454 Quality Paired end Long reads Bisulfite
reference reads suffix array
SOAP [51] X X X X
MAQ [54] X X X X X
Mosaik X X X X X
Eland X X
SSAHA2 [61] X X X
Bowtie [67] X X X X
BWA [69] X X X X
BWA-SW [69] X X X X X
SOAP2 [70] X X X X X](https://image.slidesharecdn.com/20121003sequencing1-121003080655-phpapp01/85/Workshop-NGS-data-analysis-1-27-320.jpg)



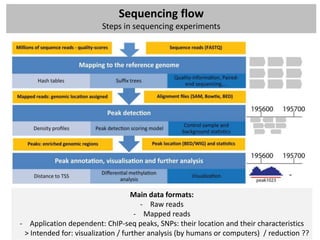
![Sequencing data formats
Other formats
- WIG files (location / annotation / scores): wiggle
Used for visulization or summarize data, in most cases count data or normalized count
data (RPKM) – extension: BigWig – binary versions (often used in GEO for ChIP-seq peaks)
browser position chr19:59304200-59310700
browser hide all
#150 base wide bar graph at arbitrarily spaced positions,
#threshold line drawn at y=11.76
#autoScale off viewing range set to [0:25]
#priority = 10 positions this as the first graph
track type=wiggle_0 name="variableStep" description="variableStep format"
visibility=full autoScale=off viewLimits=0.0:25.0 color=50,150,255
yLineMark=11.76 yLineOnOff=on priority=10
variableStep chrom=chr19 span=150
59304701 10.0
59304901 12.5
59305401 15.0
59305601 17.5
59305901 20.0
59306081 17.5](https://image.slidesharecdn.com/20121003sequencing1-121003080655-phpapp01/85/Workshop-NGS-data-analysis-1-32-320.jpg)