Download as PDF, PPTX







![SAM/BAM FILES
20
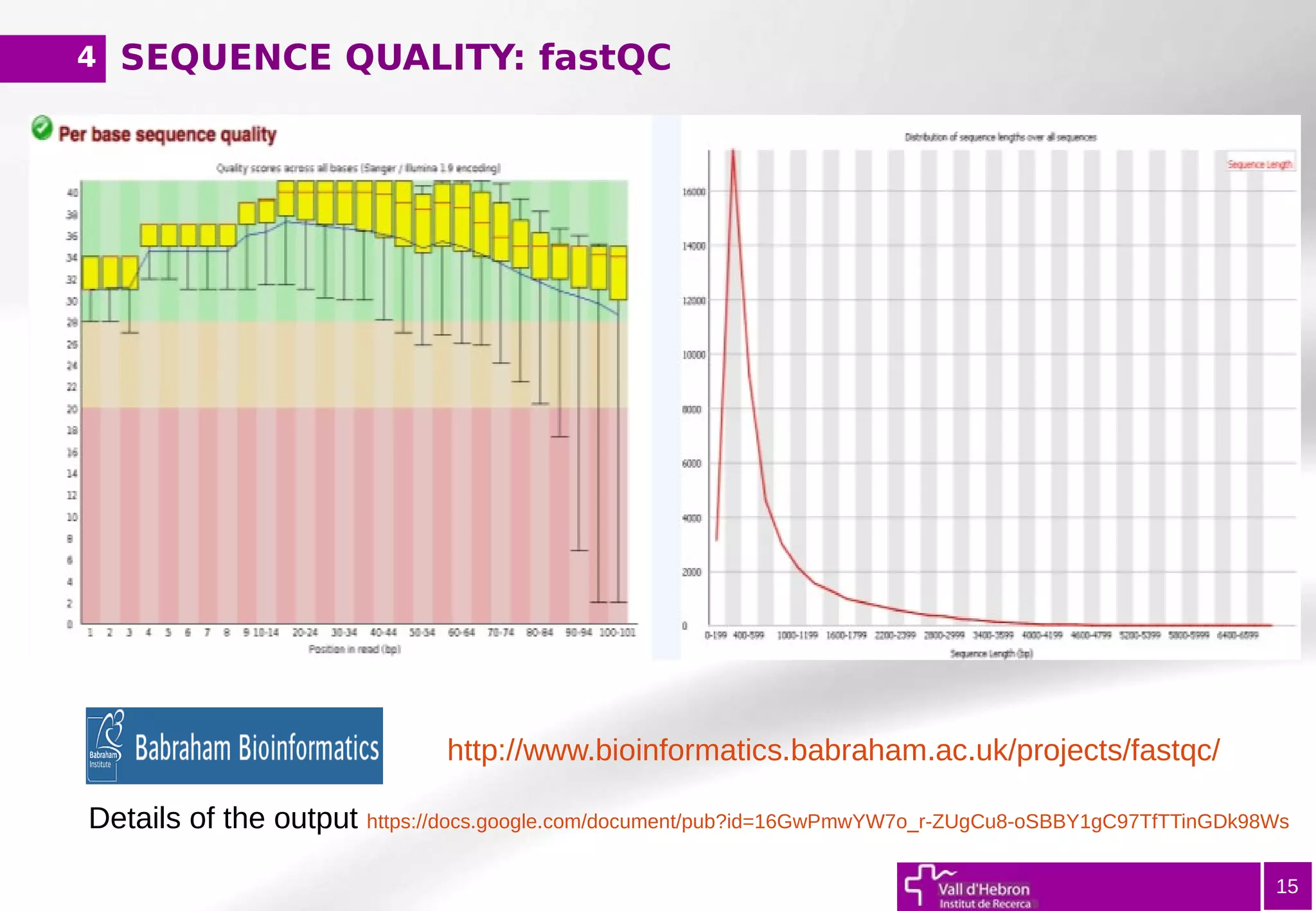
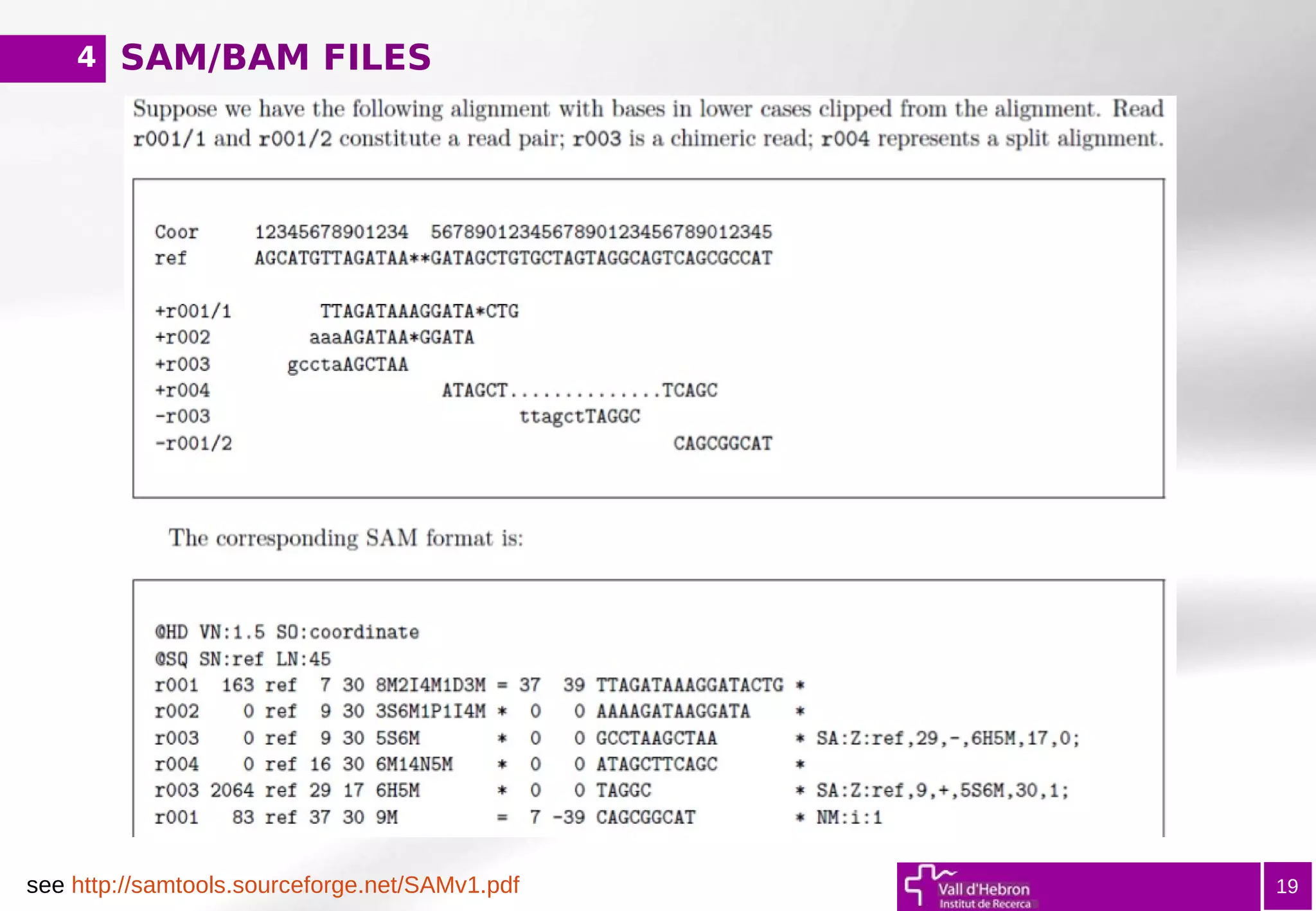
@ Header (information regarding reference genome, alignment method...)
1) Read ID (QNAME)
2) Bitwise FLAG (first/second read in pair, both reads mapped...)
3) ReferenceSequence Name (RNAME)
4) Position (POS, coordinate)
5) MapQuality (MAPQ = -10log10P[wrong mapping position])
6) CIGAR (describes alignment – matches, skipped regions, insertions..)
7) ReferenceSequence (RNEXT, Ref seq of the pair)
8) Position of the pair (PNEXT)
9) TemplateLength (TLEN)
10) ReadSequence
11) QUAL (in Fastq format, '*' if NA)
...
4](https://image.slidesharecdn.com/bbr-2-140612014222-phpapp02/75/Introduction-to-NGS-Variant-Calling-Analysis-UEB-UAT-Bioinformatics-Course-Session-2-3-VHIR-Barcelona-20-2048.jpg)

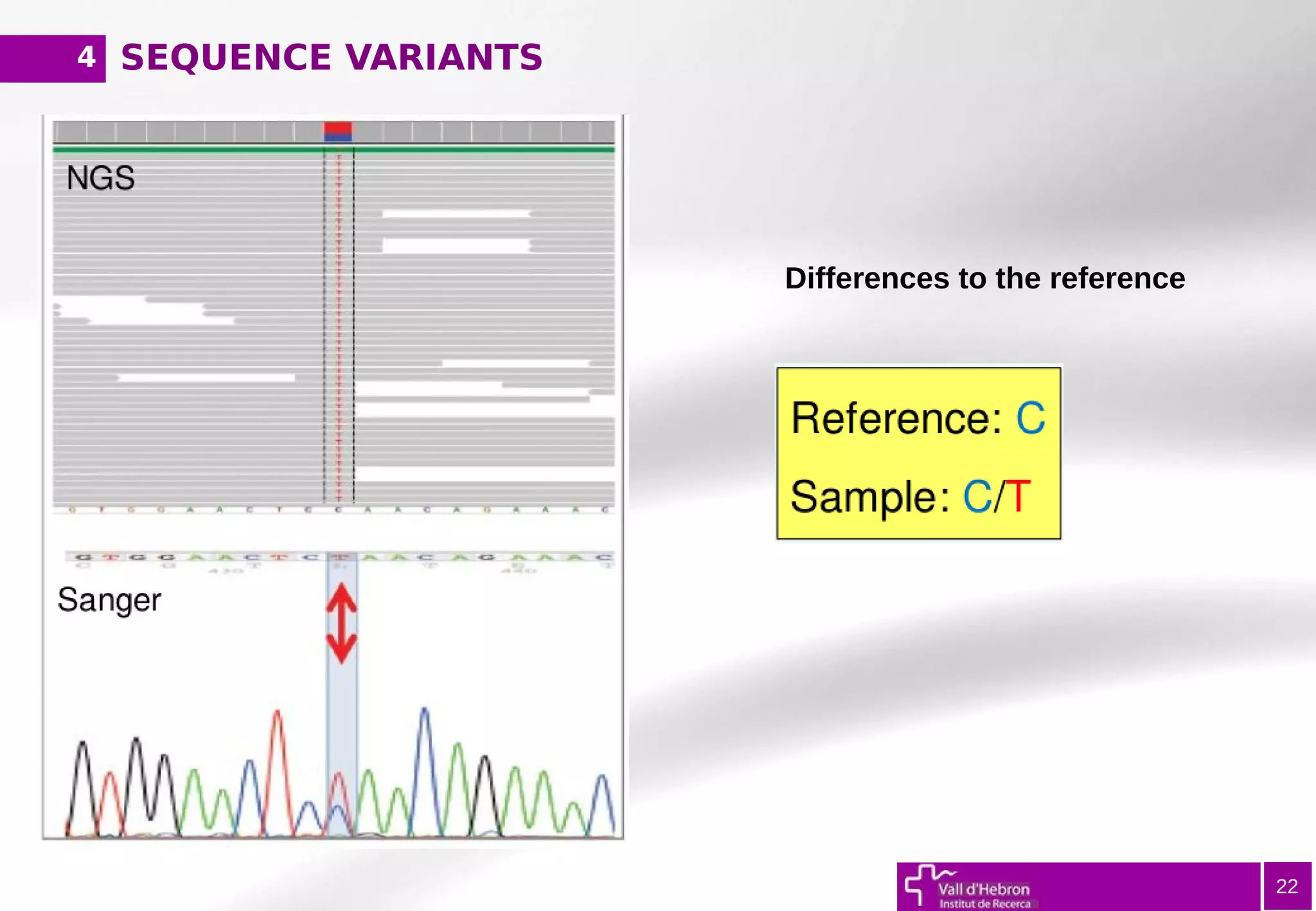
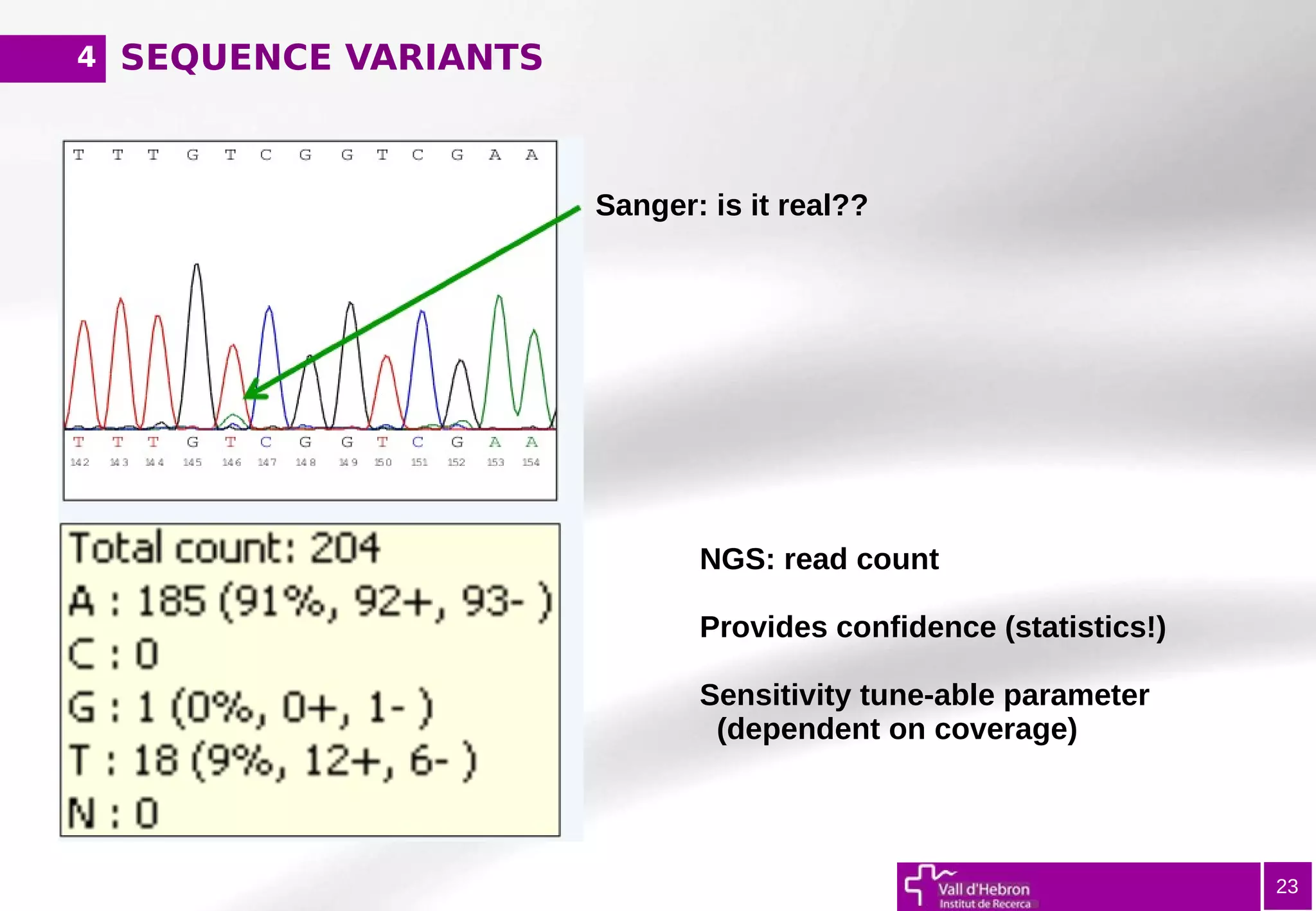

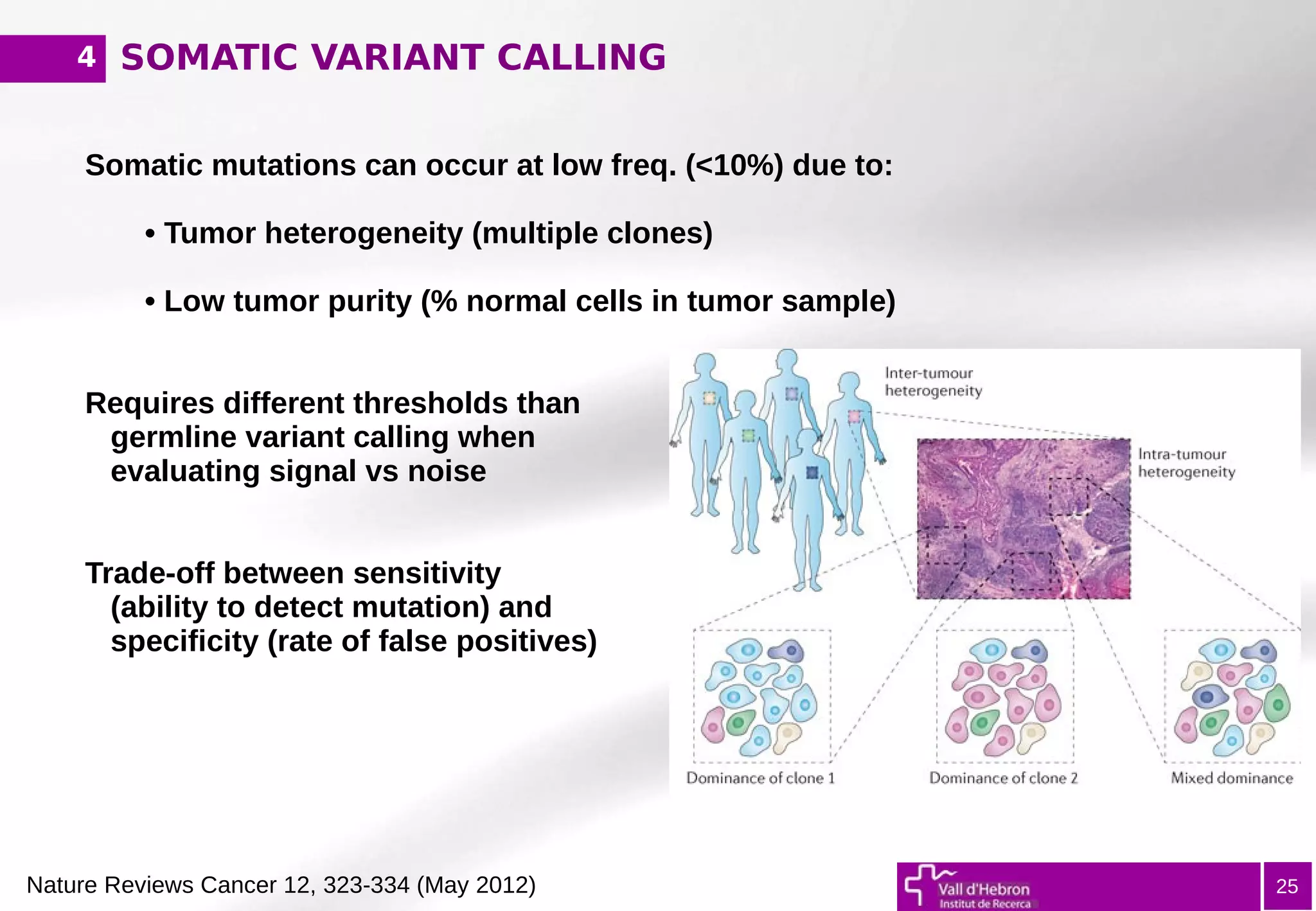



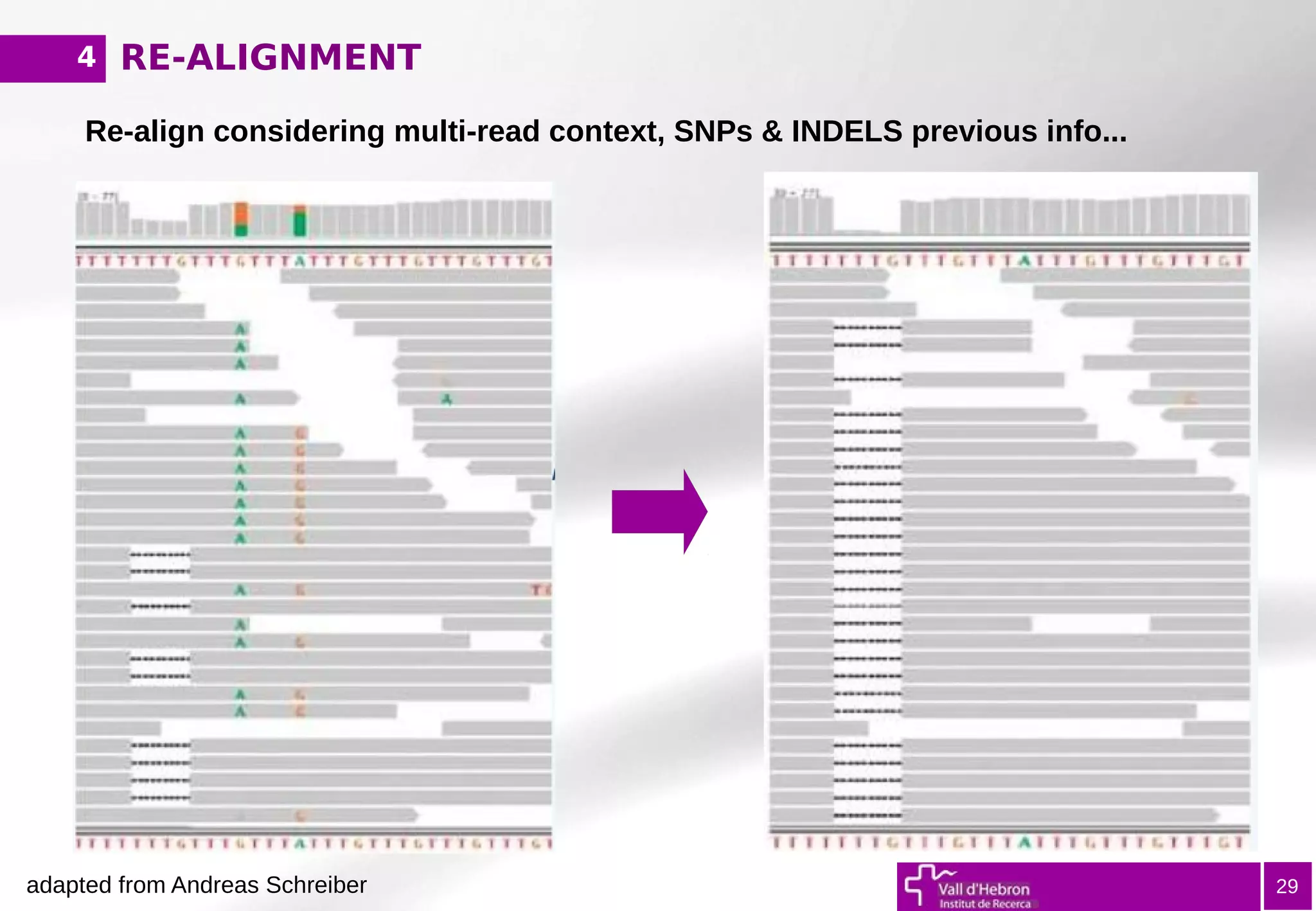


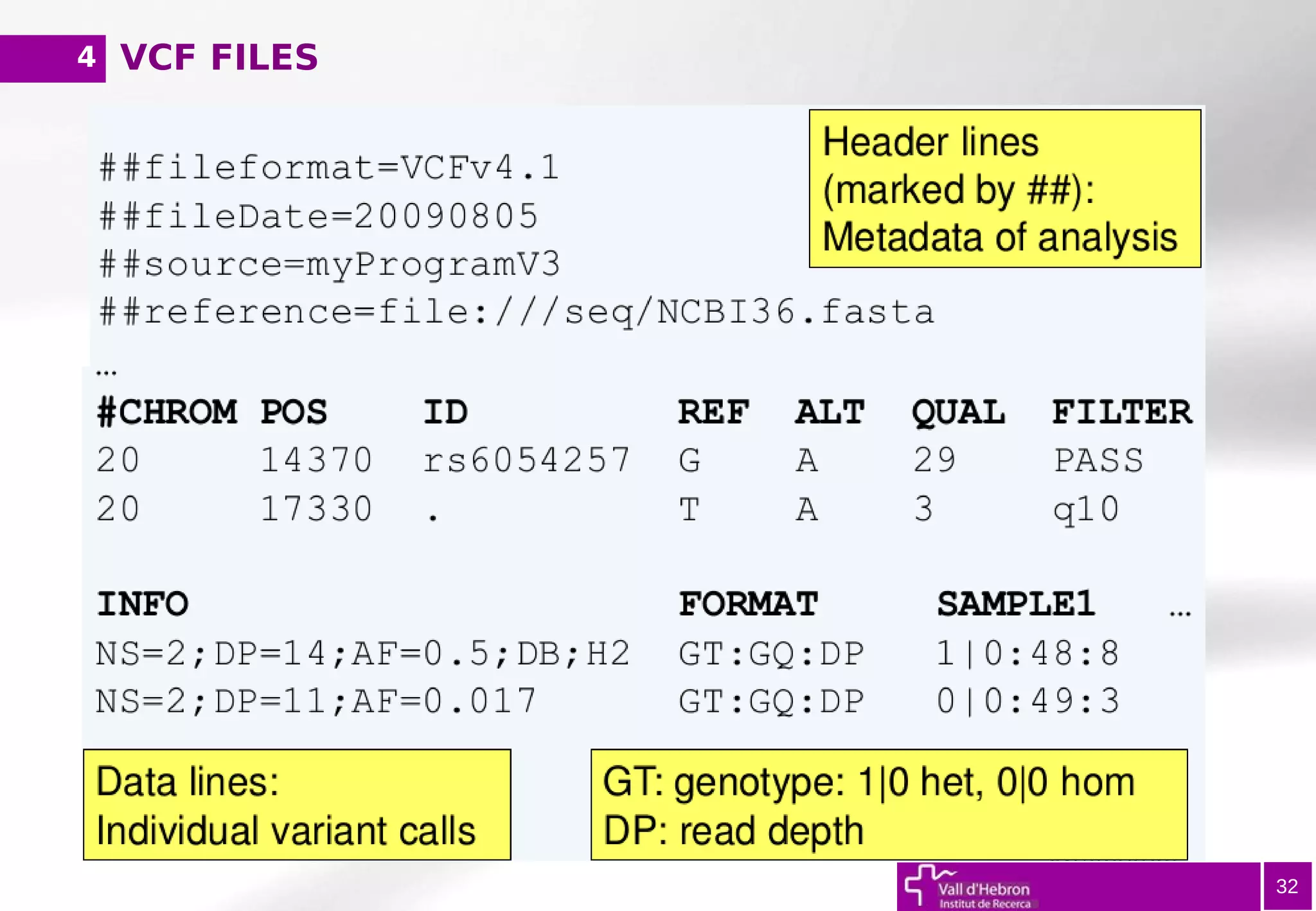
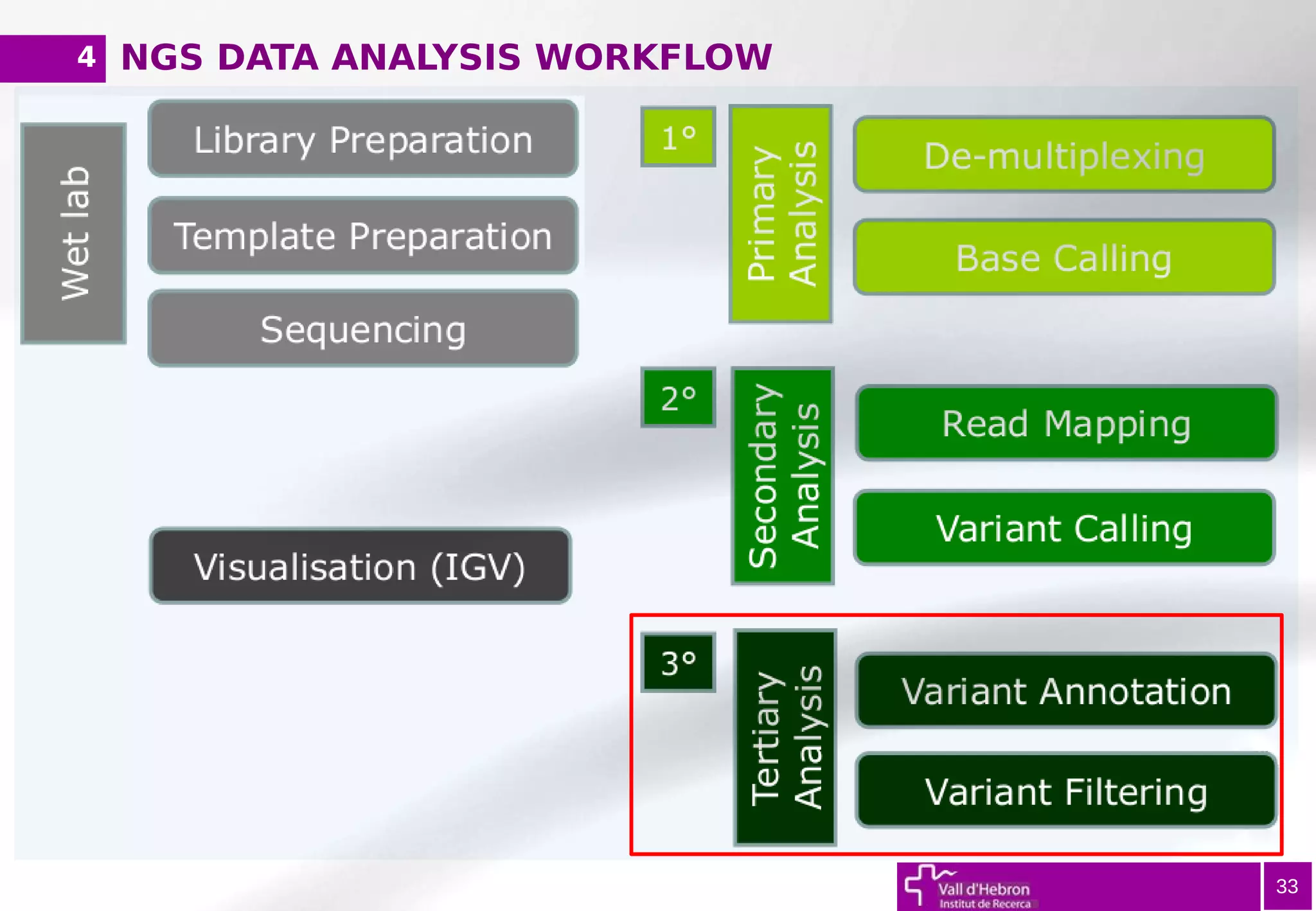

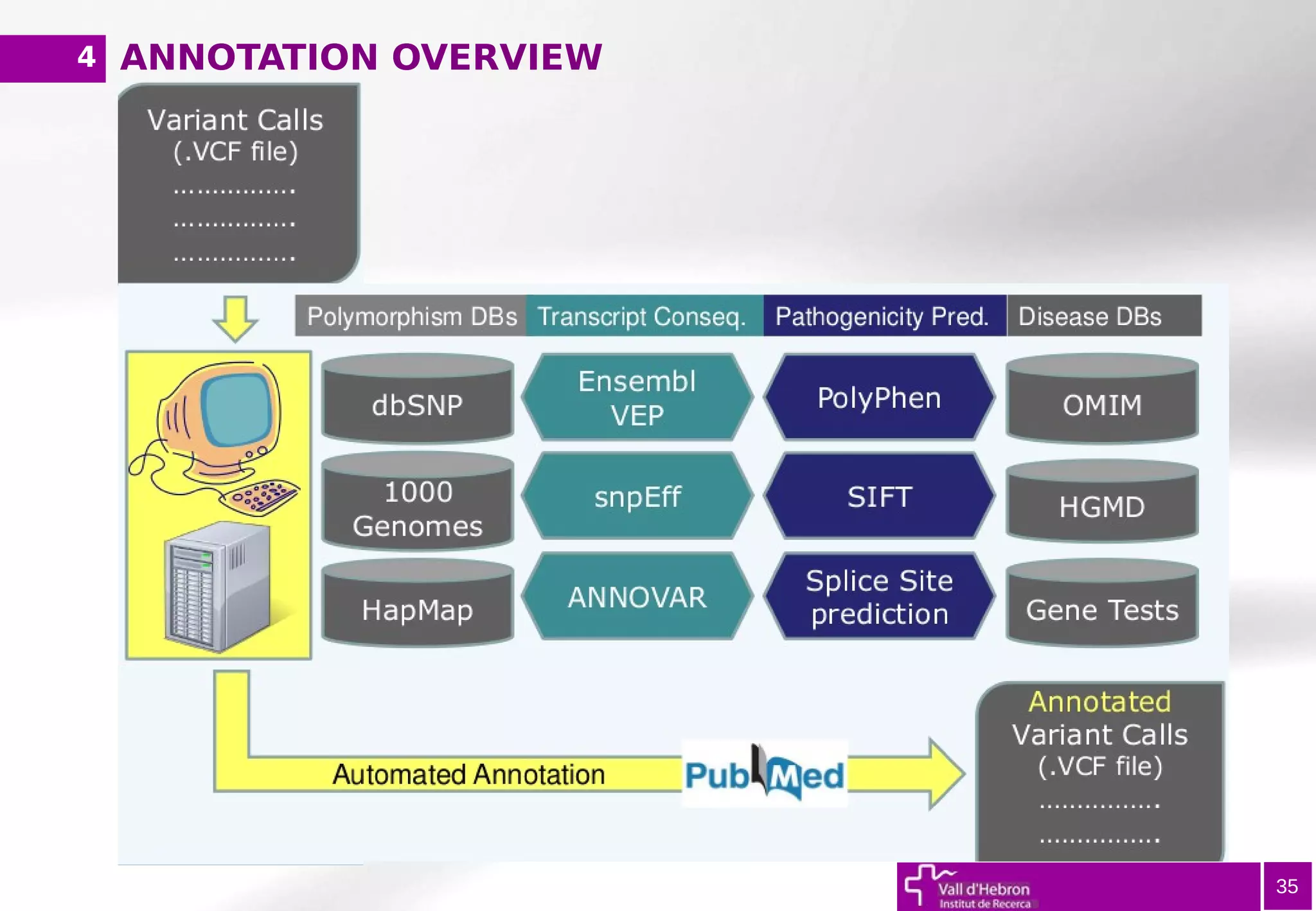
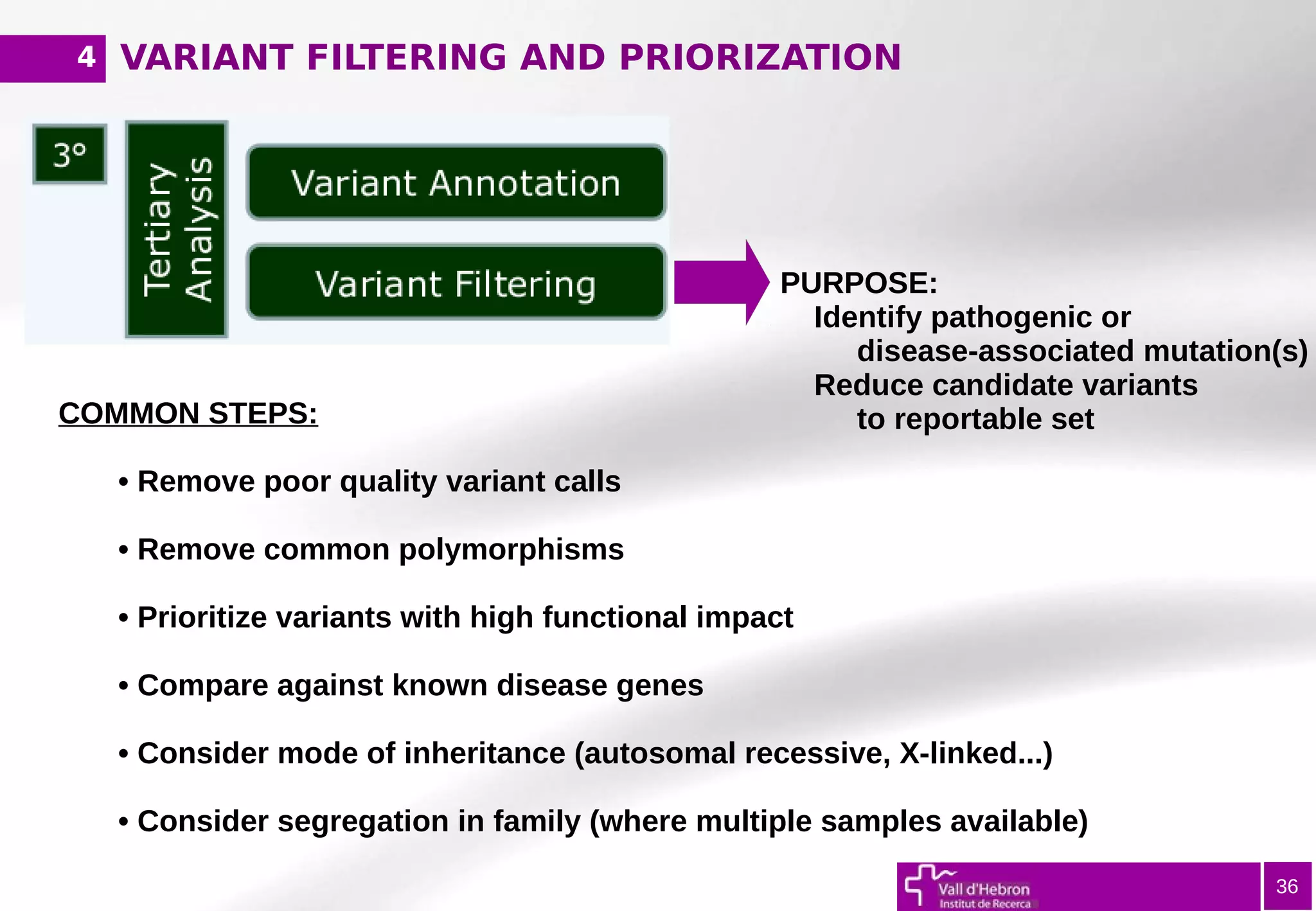
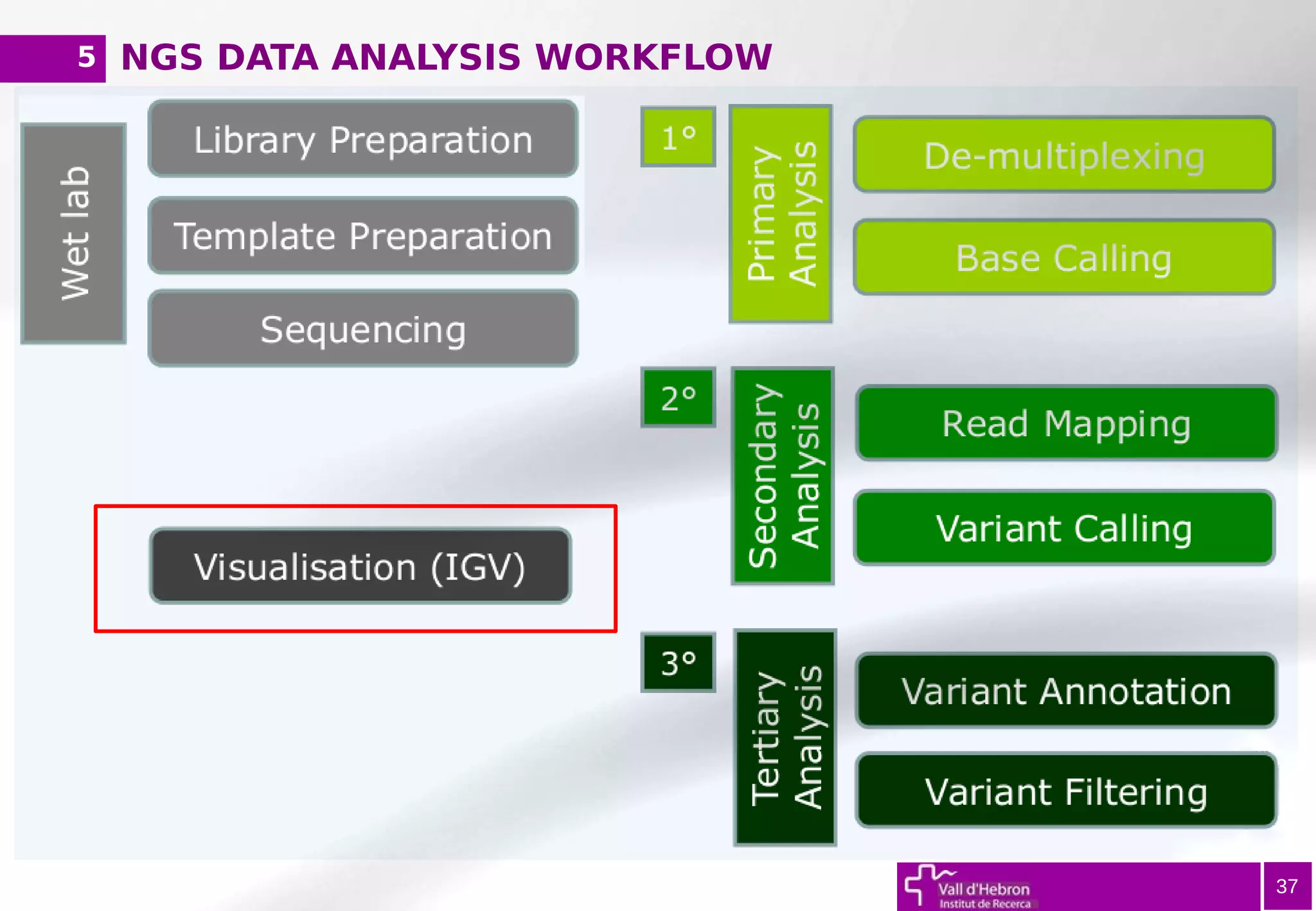
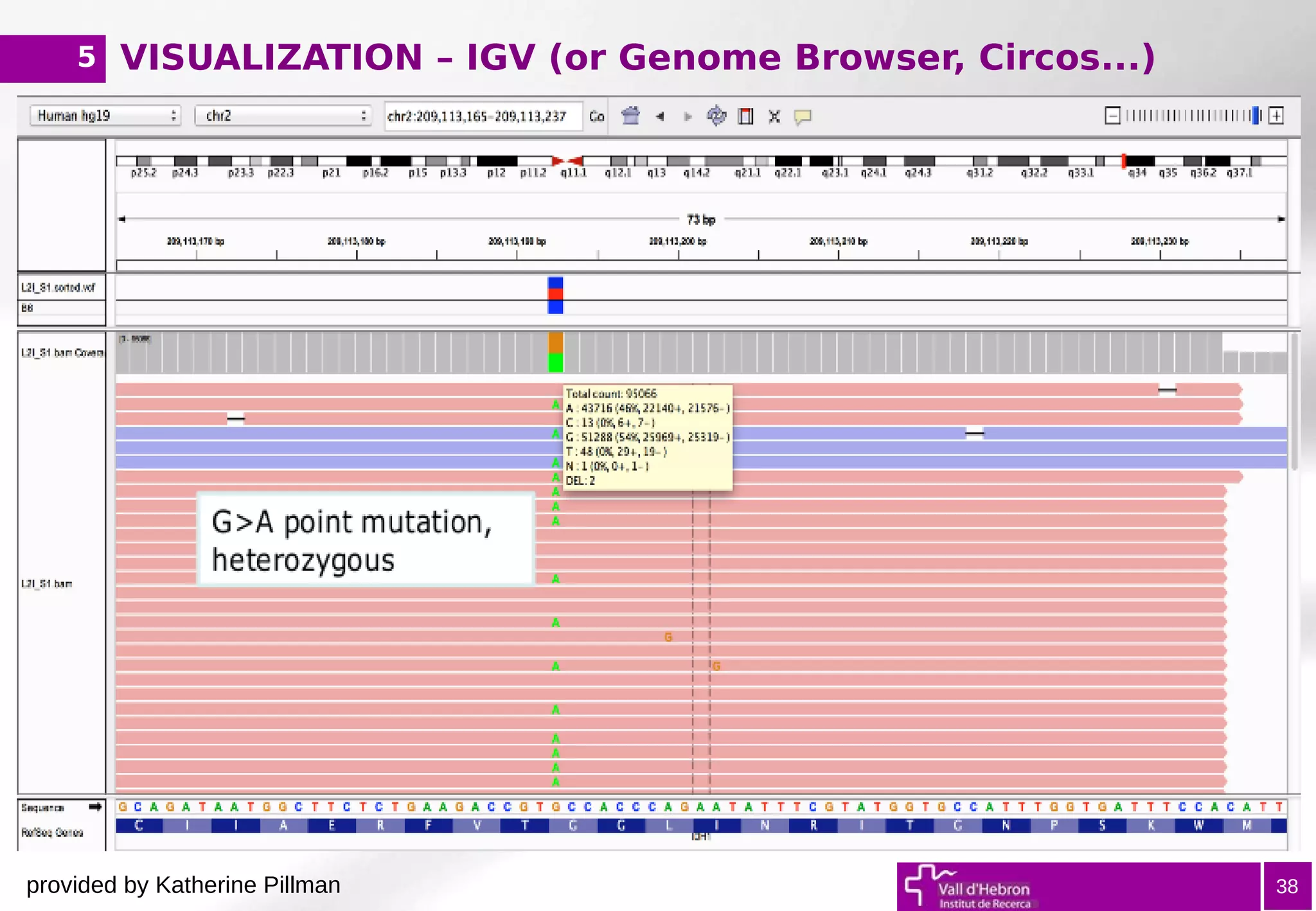
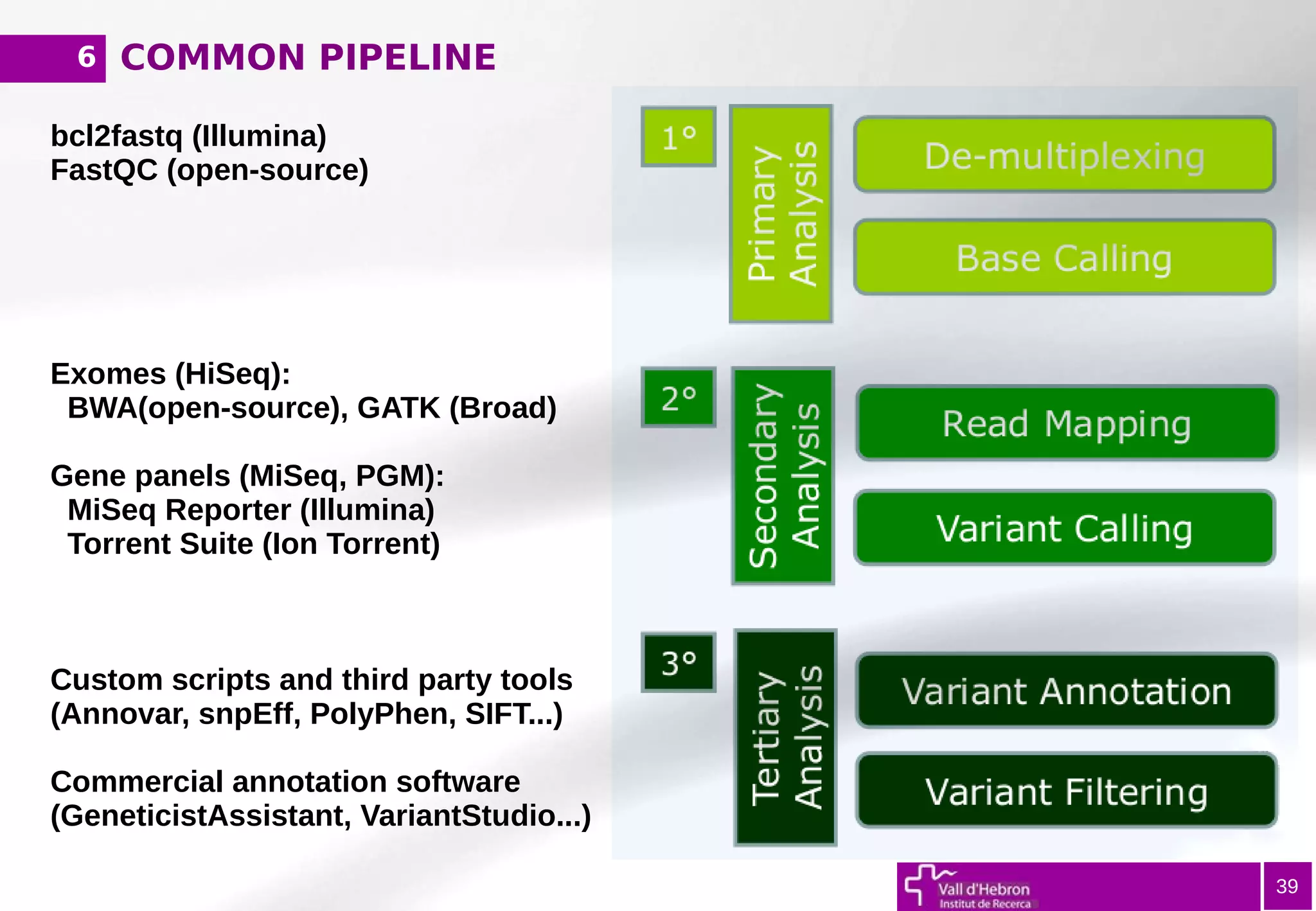
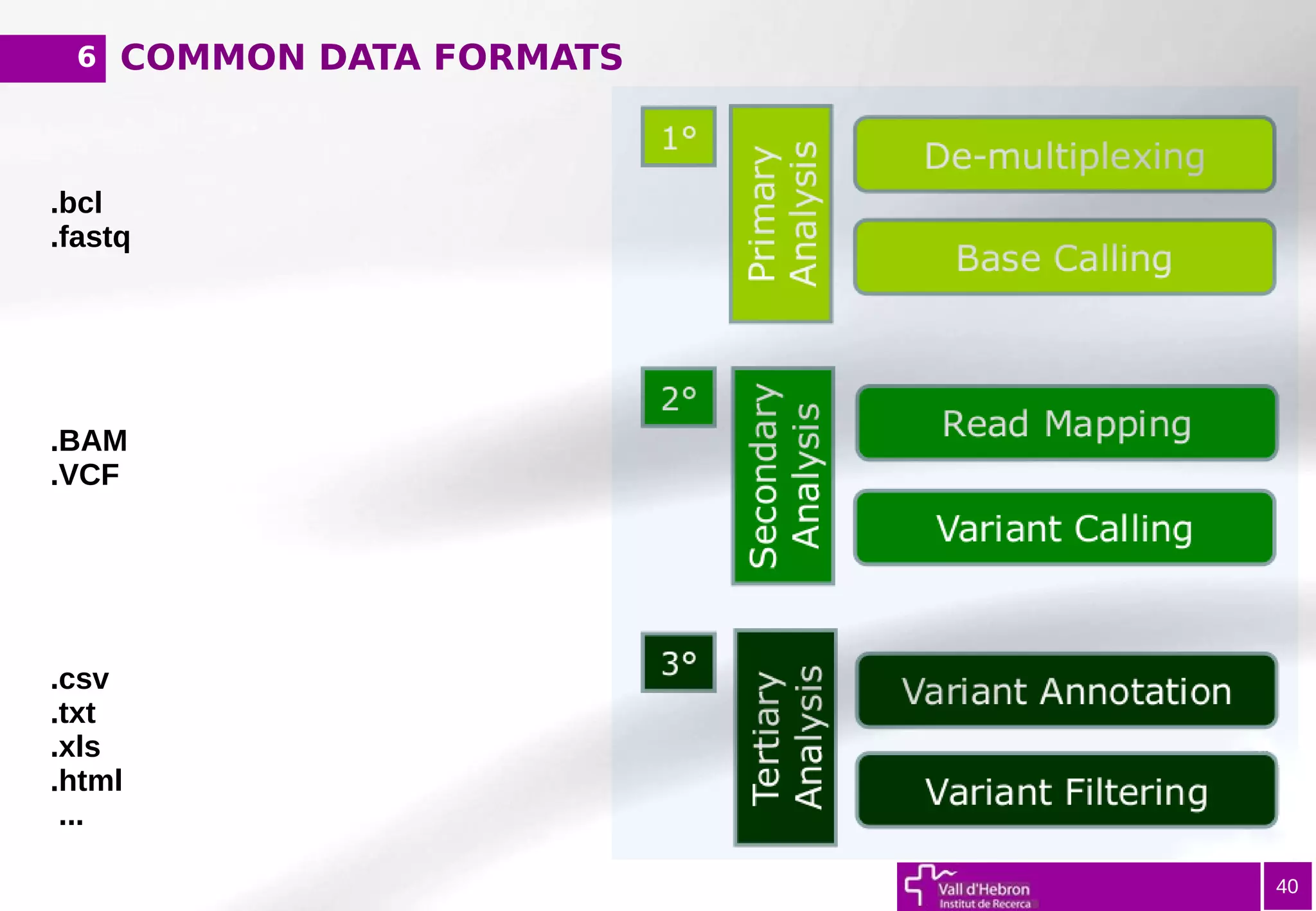


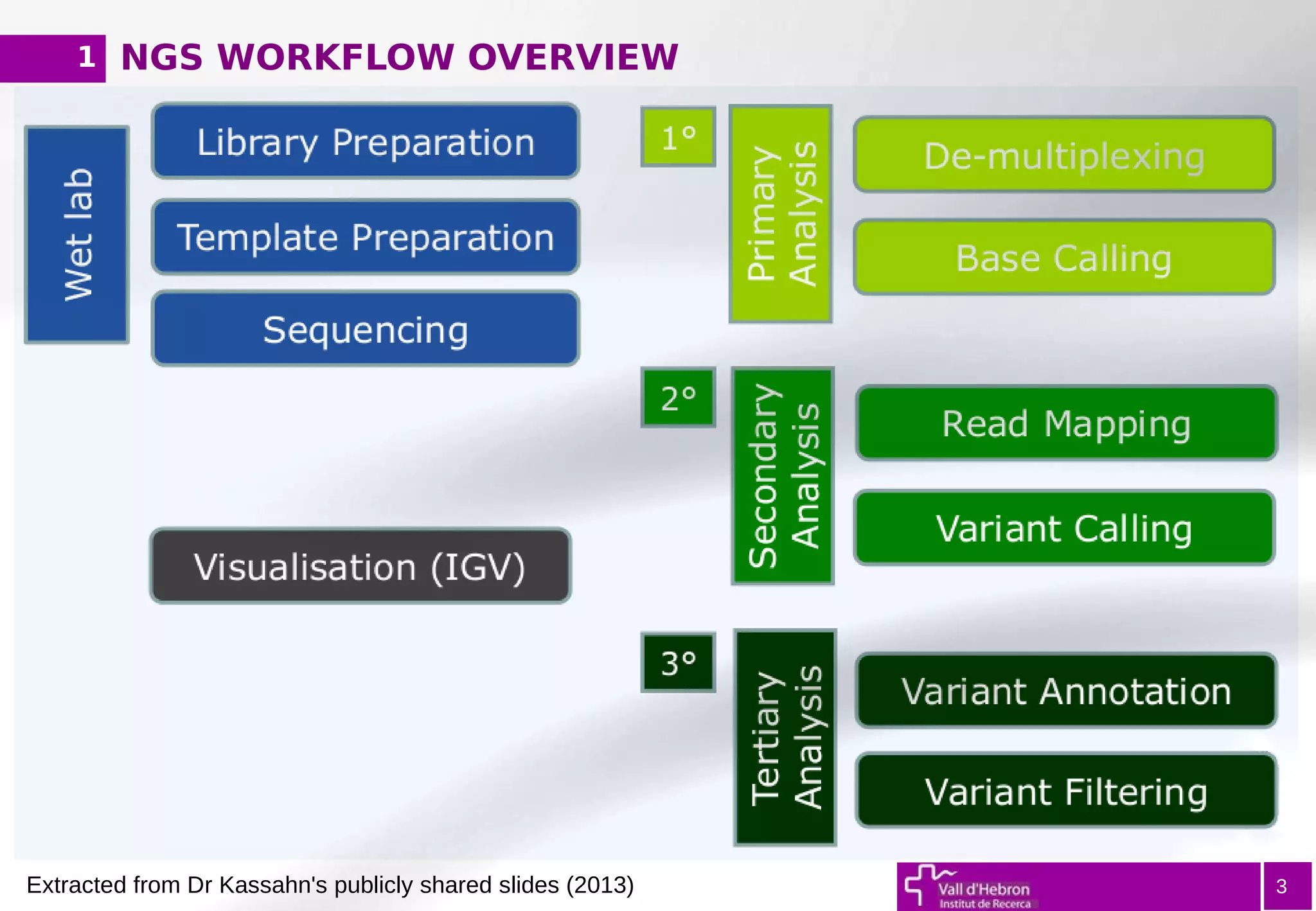
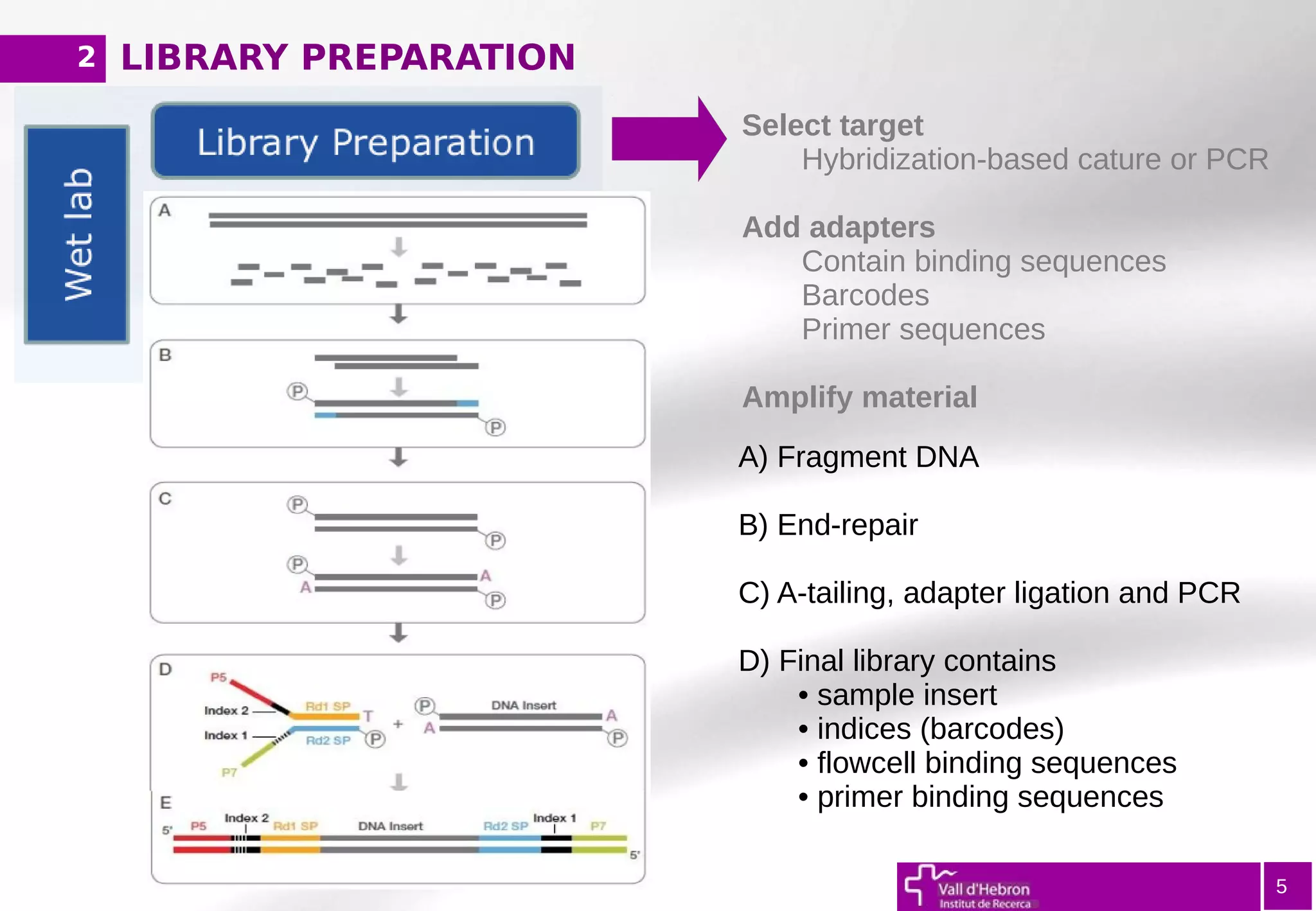
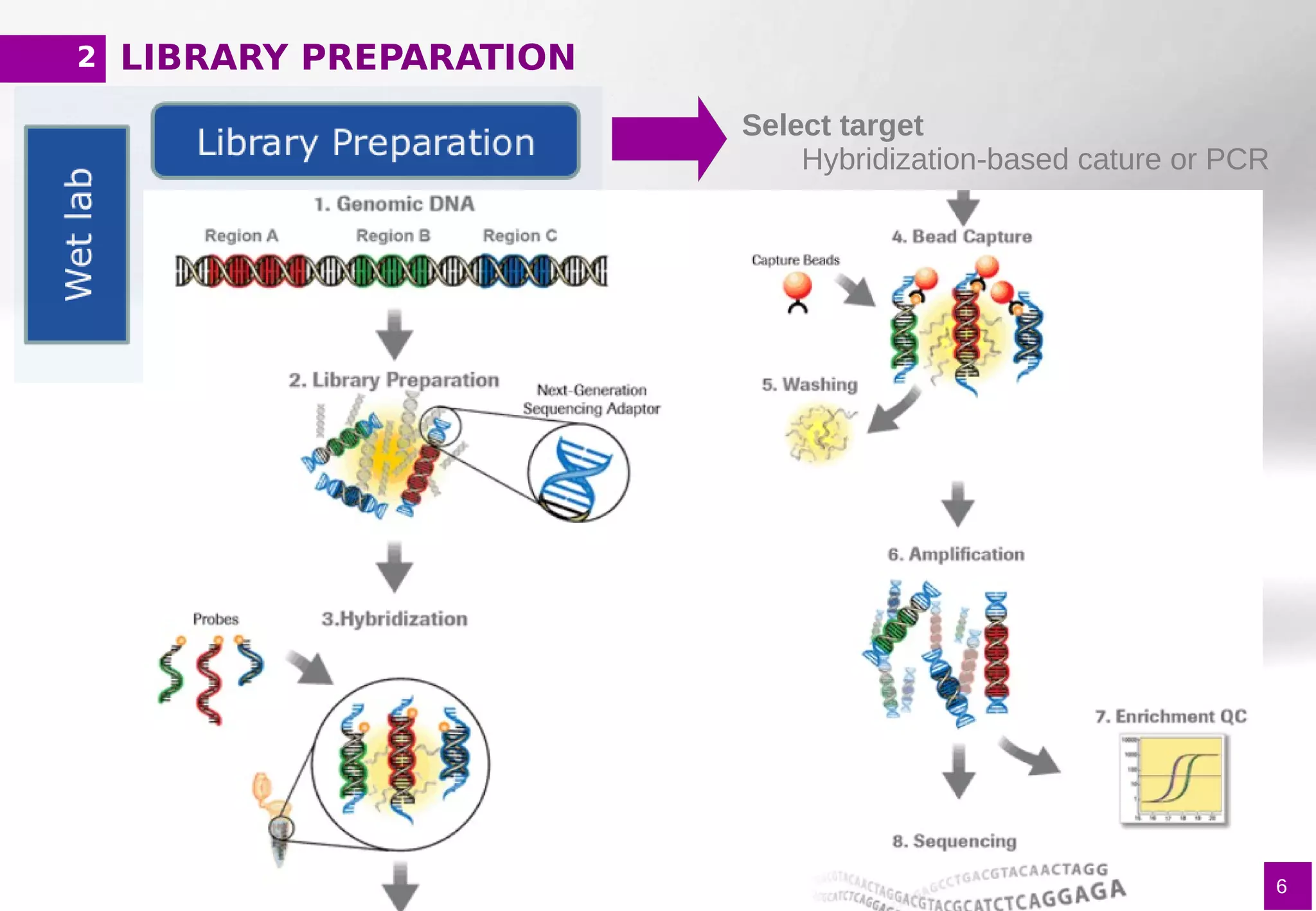
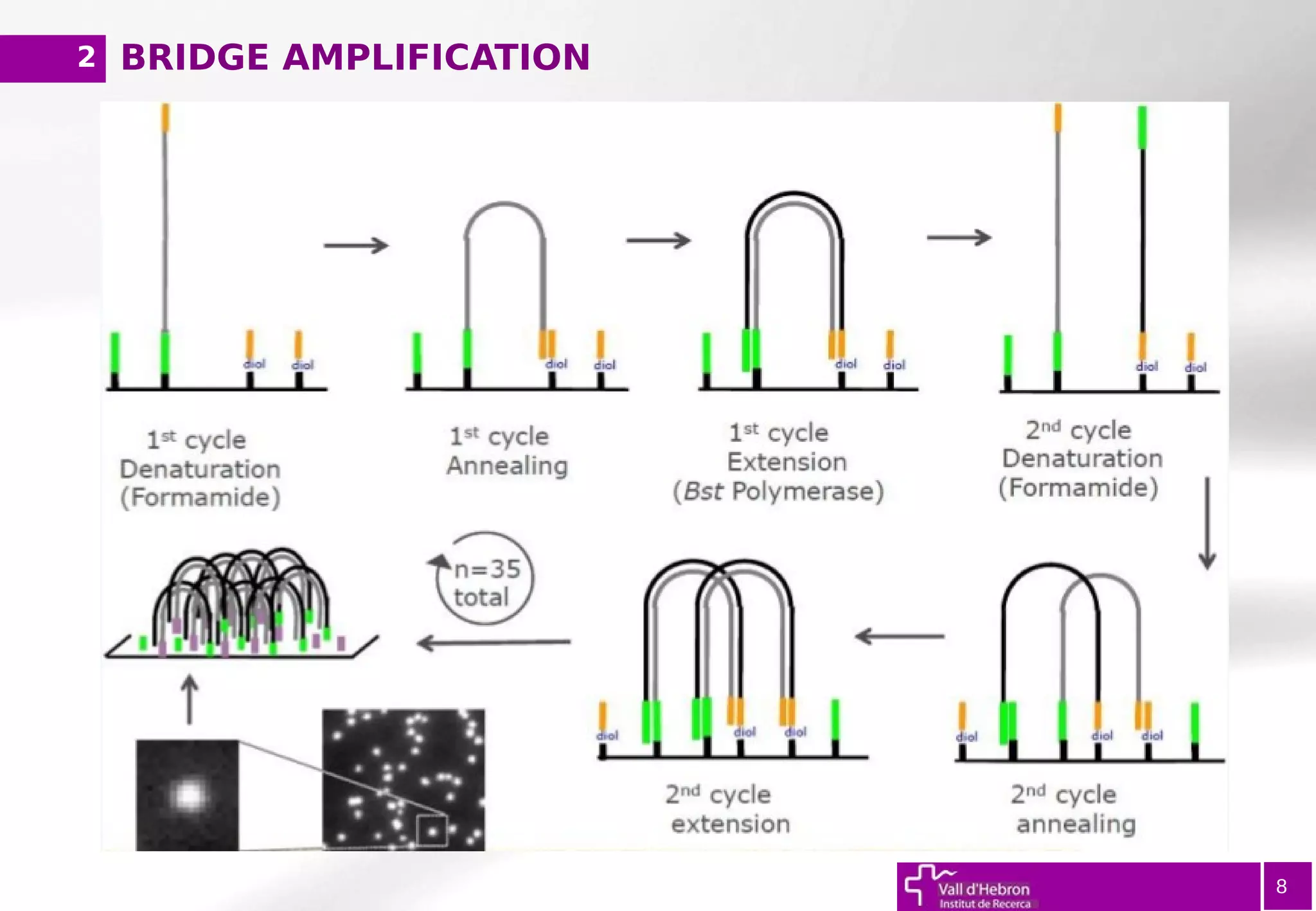
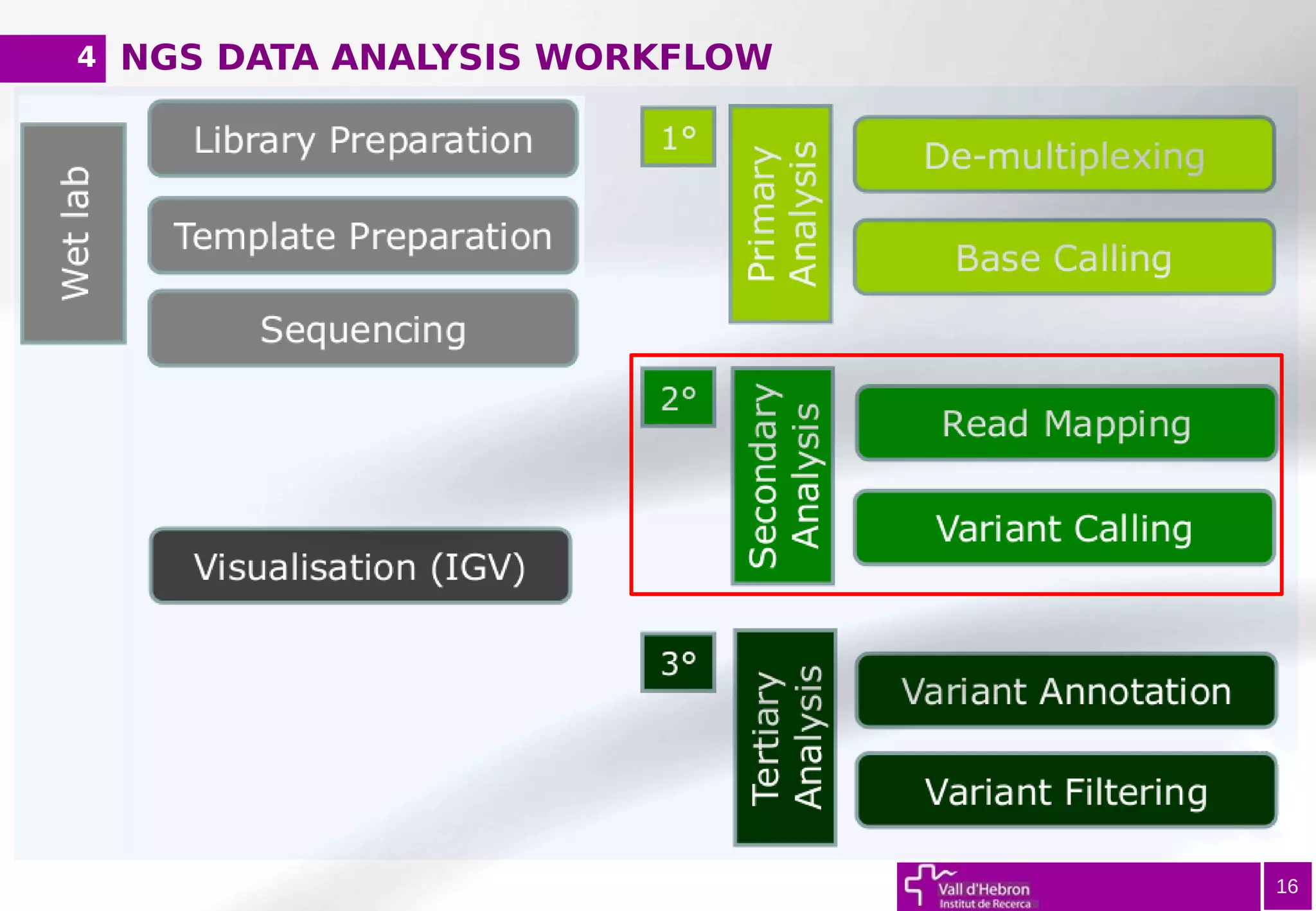
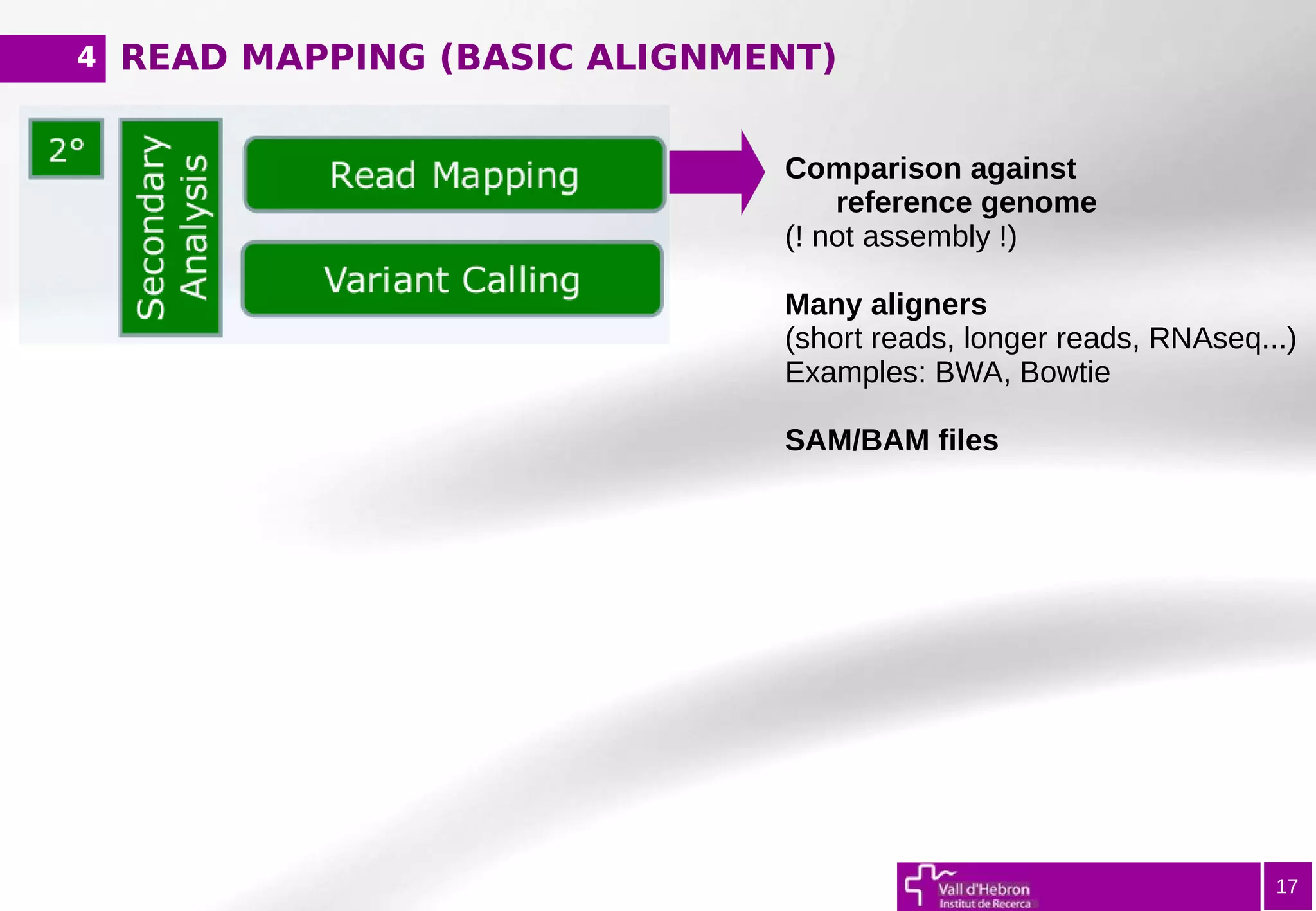
The document provides an overview of next-generation sequencing (NGS) variant calling analysis, detailing steps from library preparation to data analysis workflows. It covers critical aspects such as read mapping, variant identification, annotation, and filtering, while highlighting the complexity and variability of NGS data analysis. The conclusion emphasizes the rapid evolution of sequencing technologies and the need for standardization in bioinformatics pipelines.























































































