Downloaded 55 times




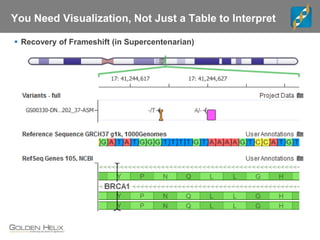

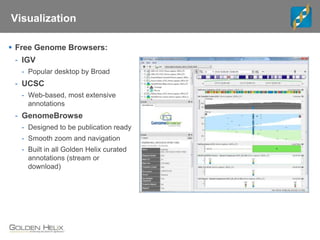

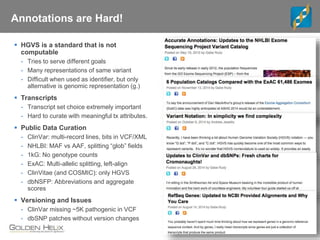



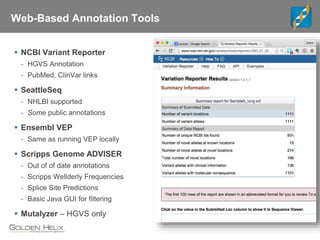
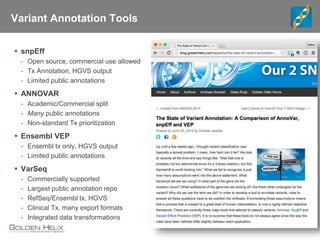



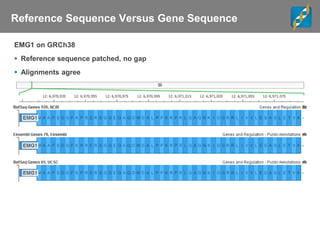
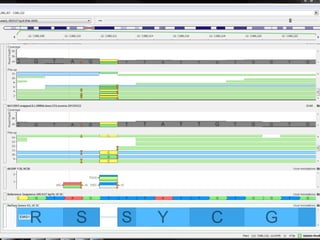

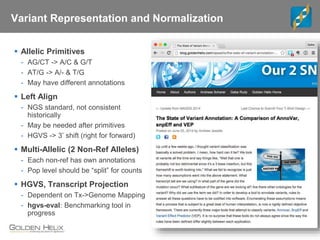

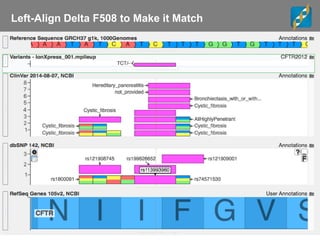
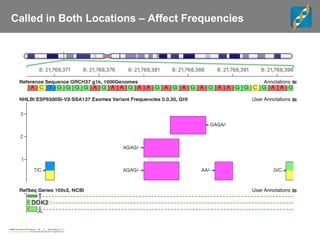
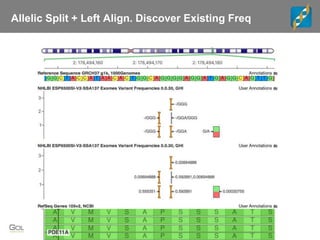
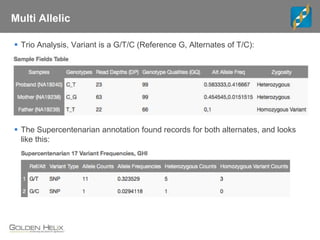
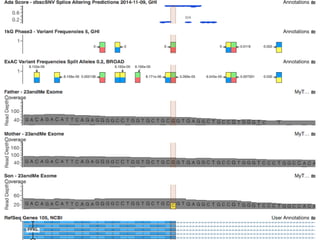

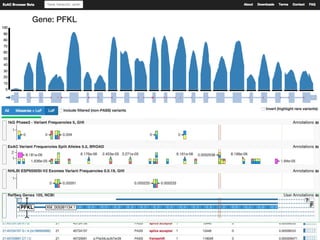
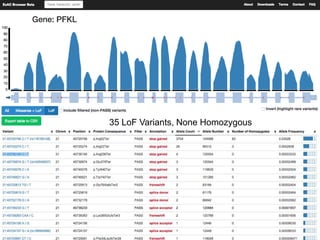


The document discusses tools and public data for variant annotation and interpretation in functional genomics. Key topics include the ACMG guidelines for variant classification, visualization of variants, and the importance of accurate gene annotations and public databases. It emphasizes the need for effective variant representation and the integration of public data to support clinical decisions in genetics.

![ACMG guidelines 2015: How to interpret DNA variants? [Today's paper]](https://cdn.slidesharecdn.com/ss_thumbnails/todayspaper200630nosound-200719080155-thumbnail.jpg?width=640&height=640&fit=bounds)