Downloaded 480 times

























![Therapy
Hydroxyurea
S-phase cytotoxic, myelosuppressive drug: inhibits ribonucleotide
reductase
Induces proliferation of early erythroid progenitors
Leads to ↑ Hgb F production (α2γ2)
γ subunit production α2 γS does not polymerize
Additional effects of hydroxyurea:
↓ Neutrophil numbers and neutrophil activation
↓ stress reticulocytes, ↓ reticulocyte adhesion
↓ endothelial adhesion properties (↓VCAM-1, ↓ laminin, ↓thrombospondin)
Improved RBC hydration and MCV
Increased [Hgb]](https://image.slidesharecdn.com/sicklecelldiseasesandip-130720225259-phpapp02/85/Sickle-cell-disease-sandip-35-320.jpg)







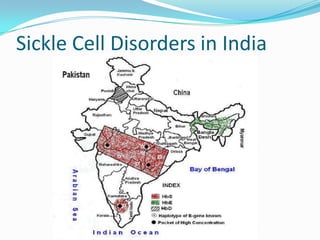
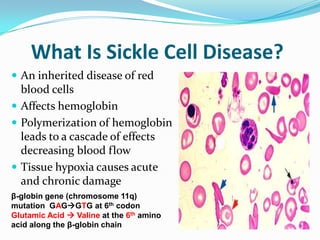
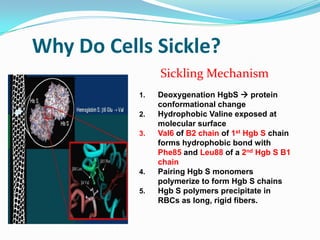
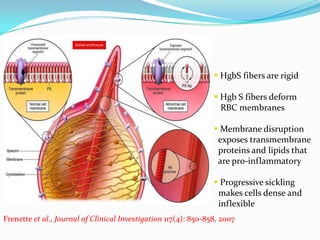
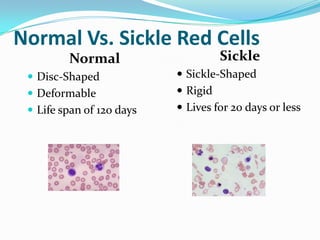
1. Sickle cell disease is an inherited blood disorder that affects hemoglobin and causes red blood cells to take on a sickle, or crescent, shape. 2. The prevalence of sickle cell disease varies by ethnicity, with the highest rates seen in African Americans at about 1 in 375 individuals. 3. Complications of sickle cell disease include anemia, infections, acute pain episodes, stroke, and damage to organs like the lungs, kidneys, spleen, and liver over time if not properly managed.

















































































