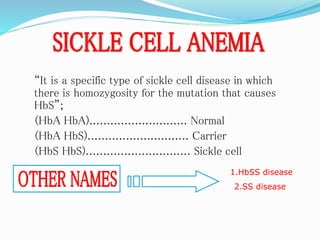
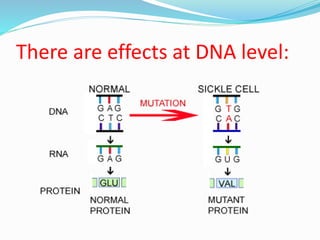
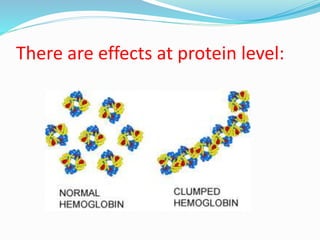
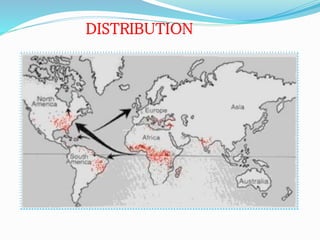
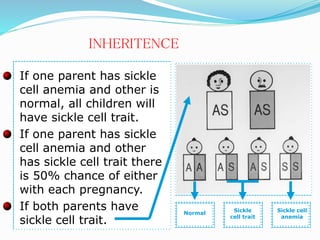
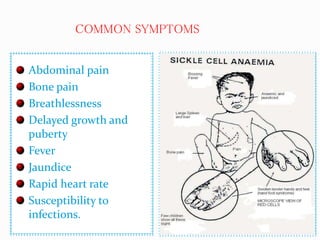
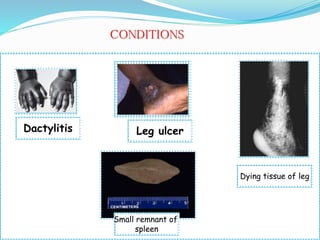
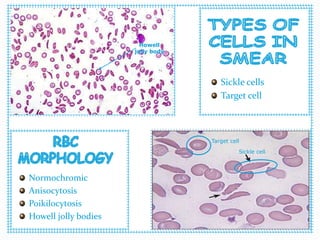
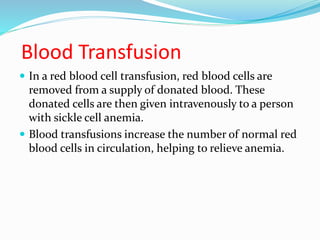
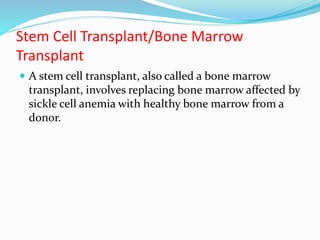
Sickle cell anemia is a genetic blood disorder caused by a mutation in the beta-globin gene of hemoglobin. It results in rigid, sickle-shaped red blood cells that can block blood vessels. The disease was first described in the early 1900s and is most common among those with ancestry from sub-Saharan Africa, South America, Central America and India. Treatment focuses on managing pain, preventing infections, and in severe cases, blood transfusions or stem cell transplants. With proper medical care, patients can live into their 40s or 50s on average.


















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)















![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)
