Downloaded 326 times




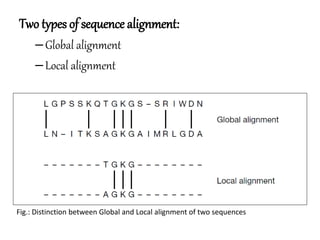





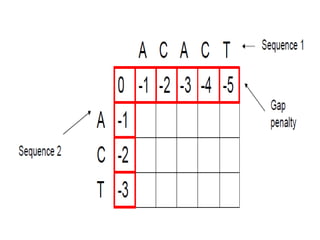

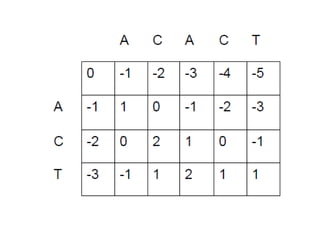

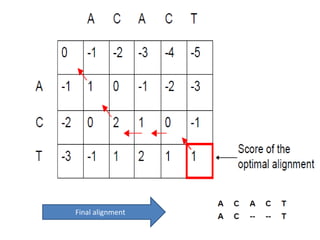




This document discusses sequence alignment and the differences between global and local alignment. It defines sequence alignment as comparing two or more sequences to find identical or similar characters in the same order. Global alignment attempts to align the entire sequences, while local alignment finds the regions of highest similarity that may only be part of the sequences. Dynamic programming is used to calculate optimal alignments through initialization of a scoring matrix, filling it, and tracing back the highest scores. The Needleman-Wunch algorithm performs global alignment, while Smith-Waterman performs local alignment by setting negative scores to zero to terminate early alignments.



































































![PSA [Conclusion]](https://cdn.slidesharecdn.com/ss_thumbnails/visualizingsequencesconclusion-100209233638-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)
































