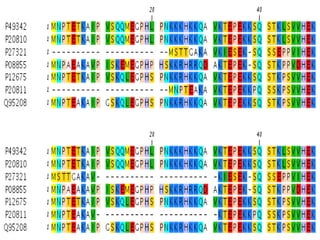
This document discusses multiple sequence alignment techniques. It begins with definitions of key terms like homology, similarity, and conservation. It then describes pairwise alignment and its applications. The rest of the document focuses on multiple sequence alignment methods like progressive alignment, iterative refinement, tree alignment, star alignment, and using genetic algorithms. It provides examples and explanations of popular multiple sequence alignment tools like Clustal W and T-Coffee.
















































































































![Human Reproduction [ Reproductive System ] Notes @irfanullah_mehar Irfanullah...](https://cdn.slidesharecdn.com/ss_thumbnails/humanreproductionreproductivesystemnotesirfanullahmeharirfanullahmeharjanantantra-260111172350-56e85778-thumbnail.jpg?width=640&height=640&fit=bounds)











