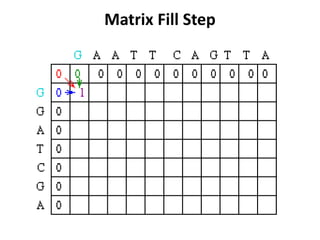
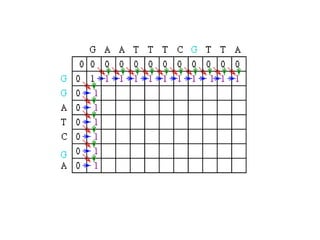
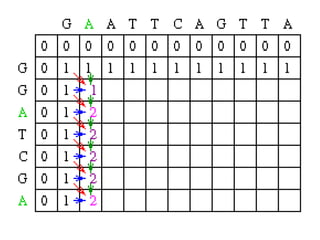
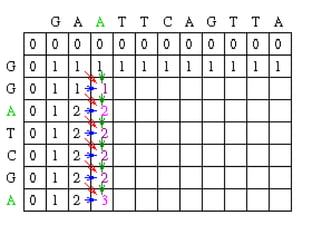
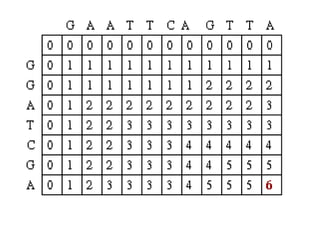
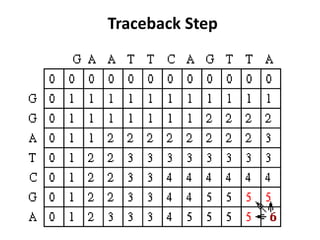
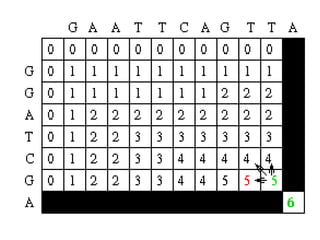
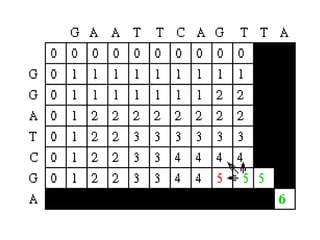
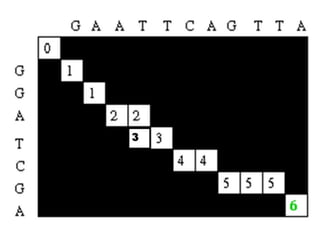
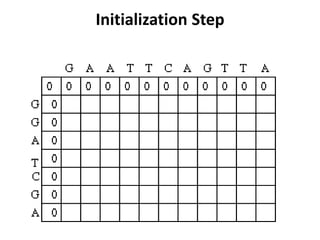
Sequence alignment involves comparing two or more sequences to identify regions of similarity. It can be used to determine if genes or proteins are evolutionarily related or identify structurally similar regions. The alignment process involves placing the sequences side by side and introducing gaps to maximize matches. Dynamic programming algorithms like Needleman-Wunsch calculate the optimal global alignment through matrix initialization, scoring, and traceback steps.


















![Scoring
• For each position, Mi,j is defined to be the
maximum score at position i,j; i.e.
Mi,j = MAXIMUM [ Mi-1, j-1 + Si,j (match/mismatch in the diagonal)
Mi,j-1 + w (gap in sequence #1),
Mi-1,j + w (gap in sequence #2)]
• In the following case, Mi-1,j-1 will be red, Mi,j-1 will
be green and Mi-1,j will be blue.](https://image.slidesharecdn.com/seqalignment-220812051104-27218e2c/85/seq-alignment-ppt-19-320.jpg)