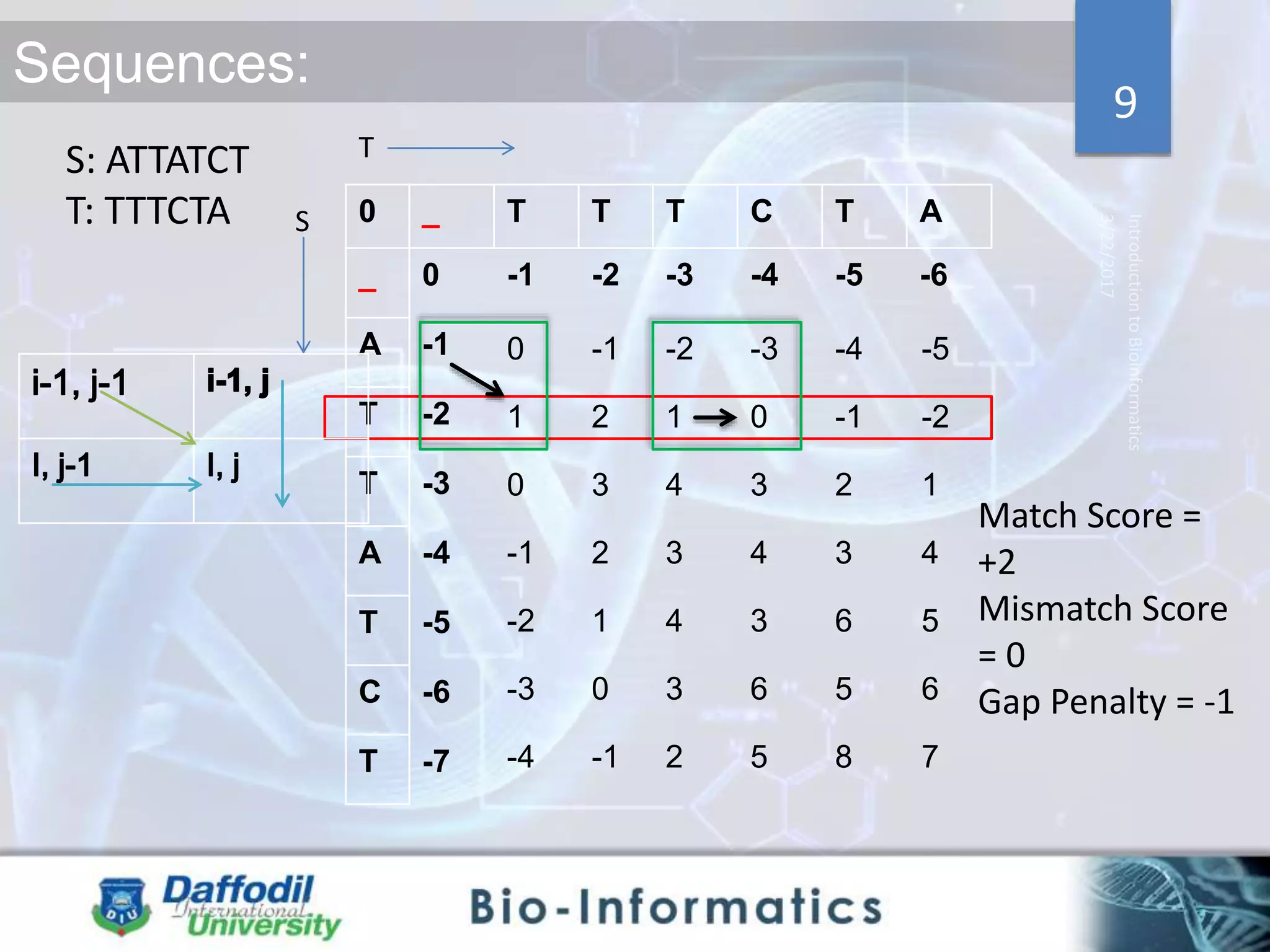
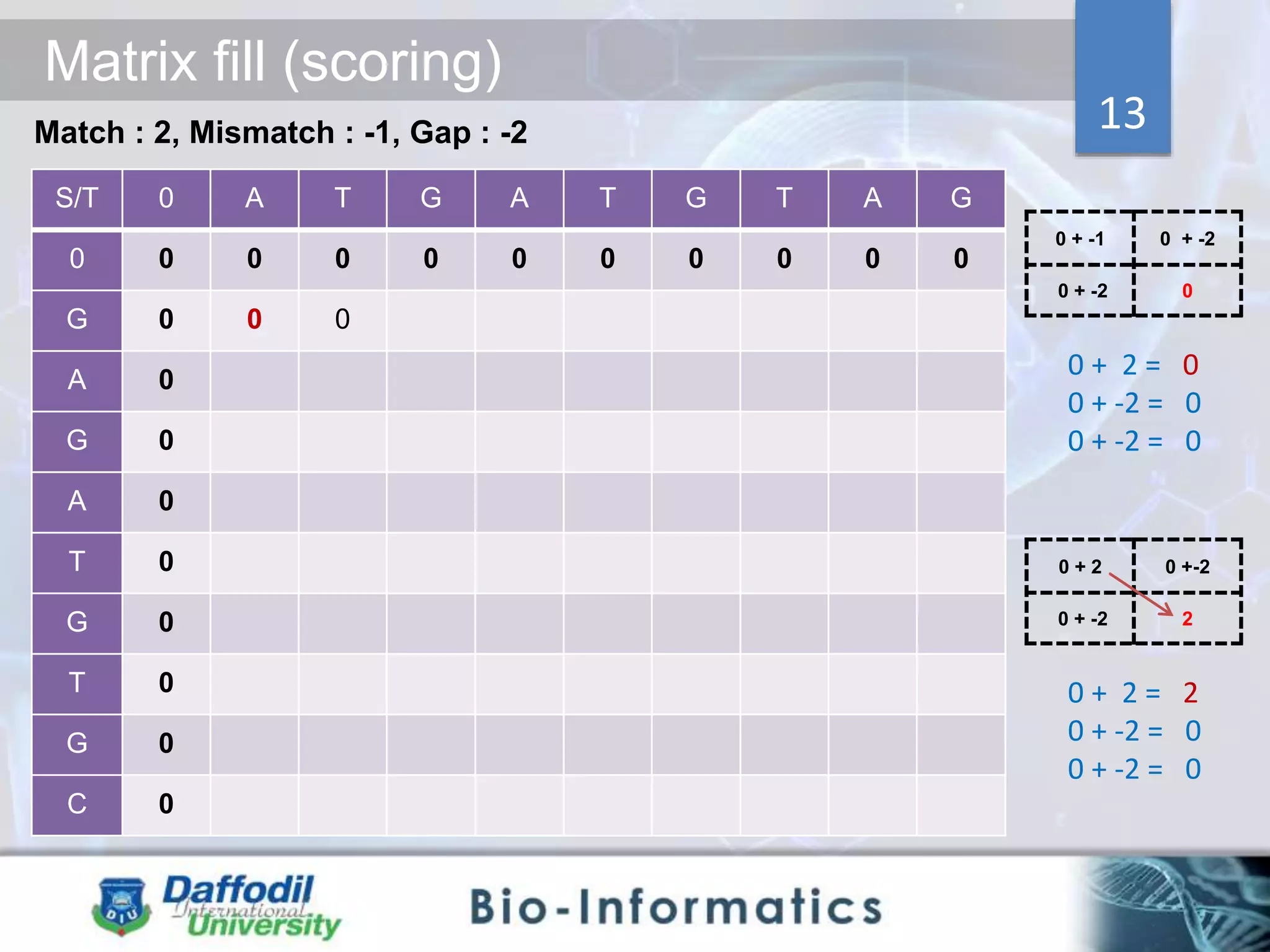
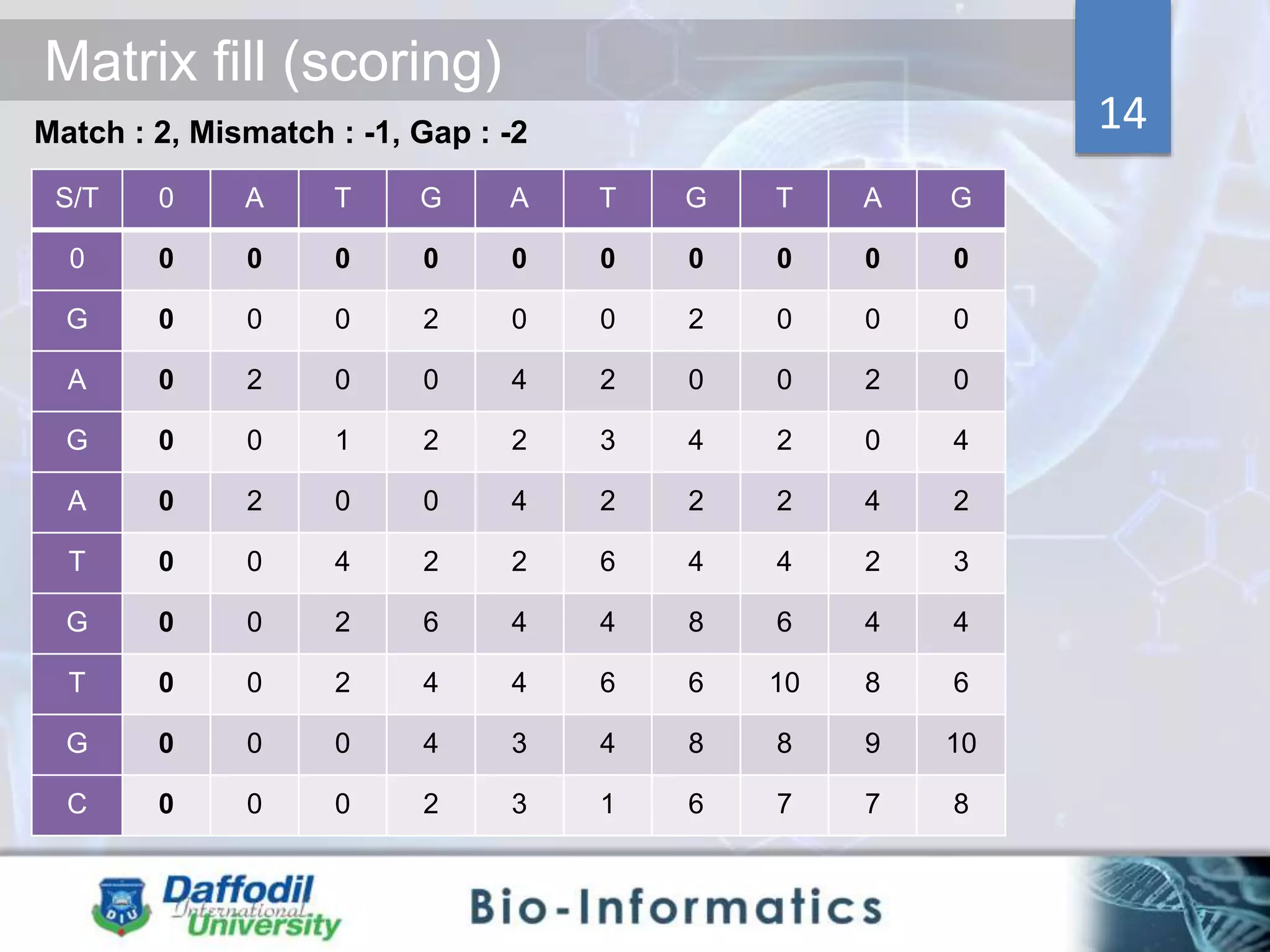
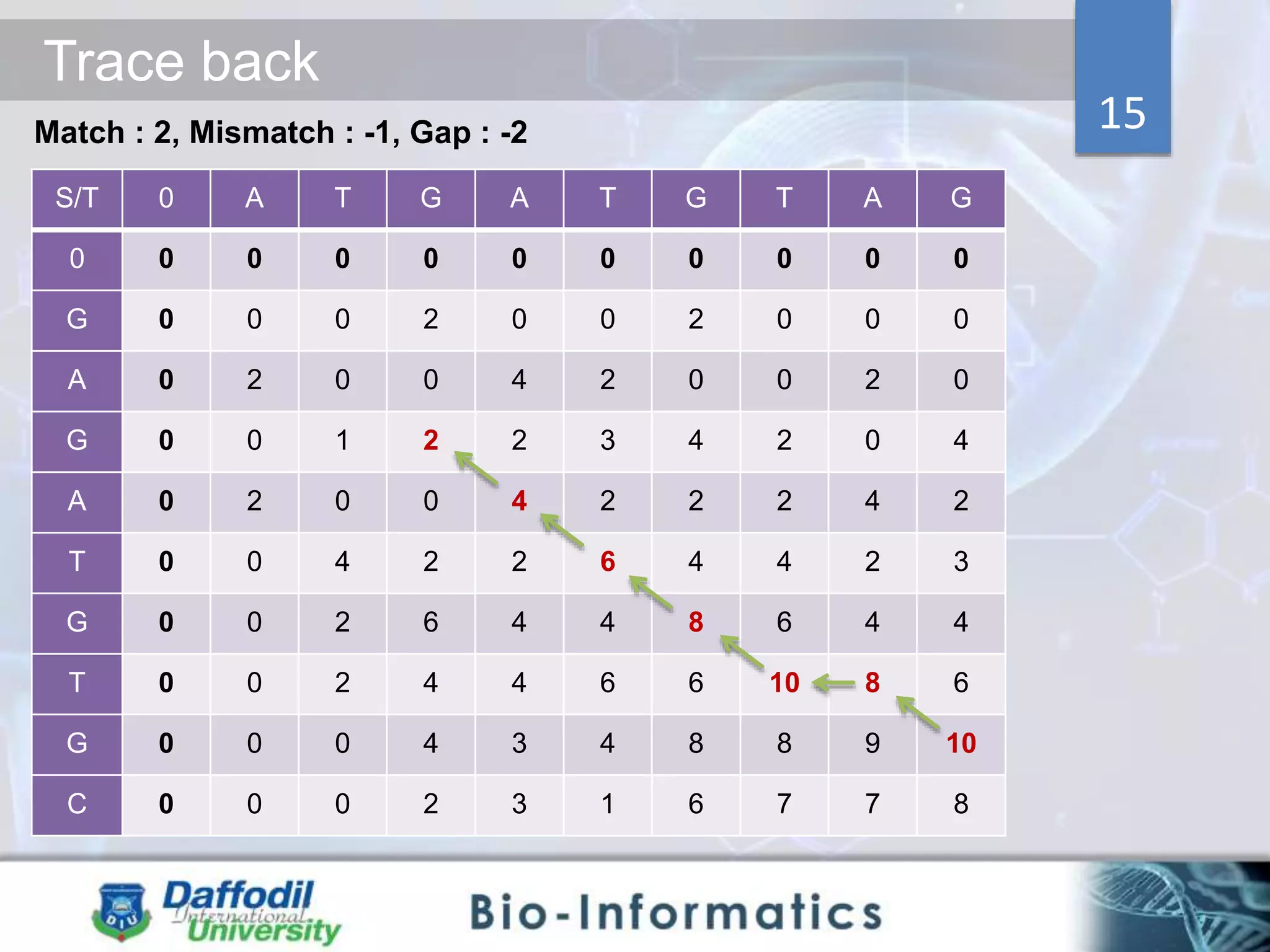
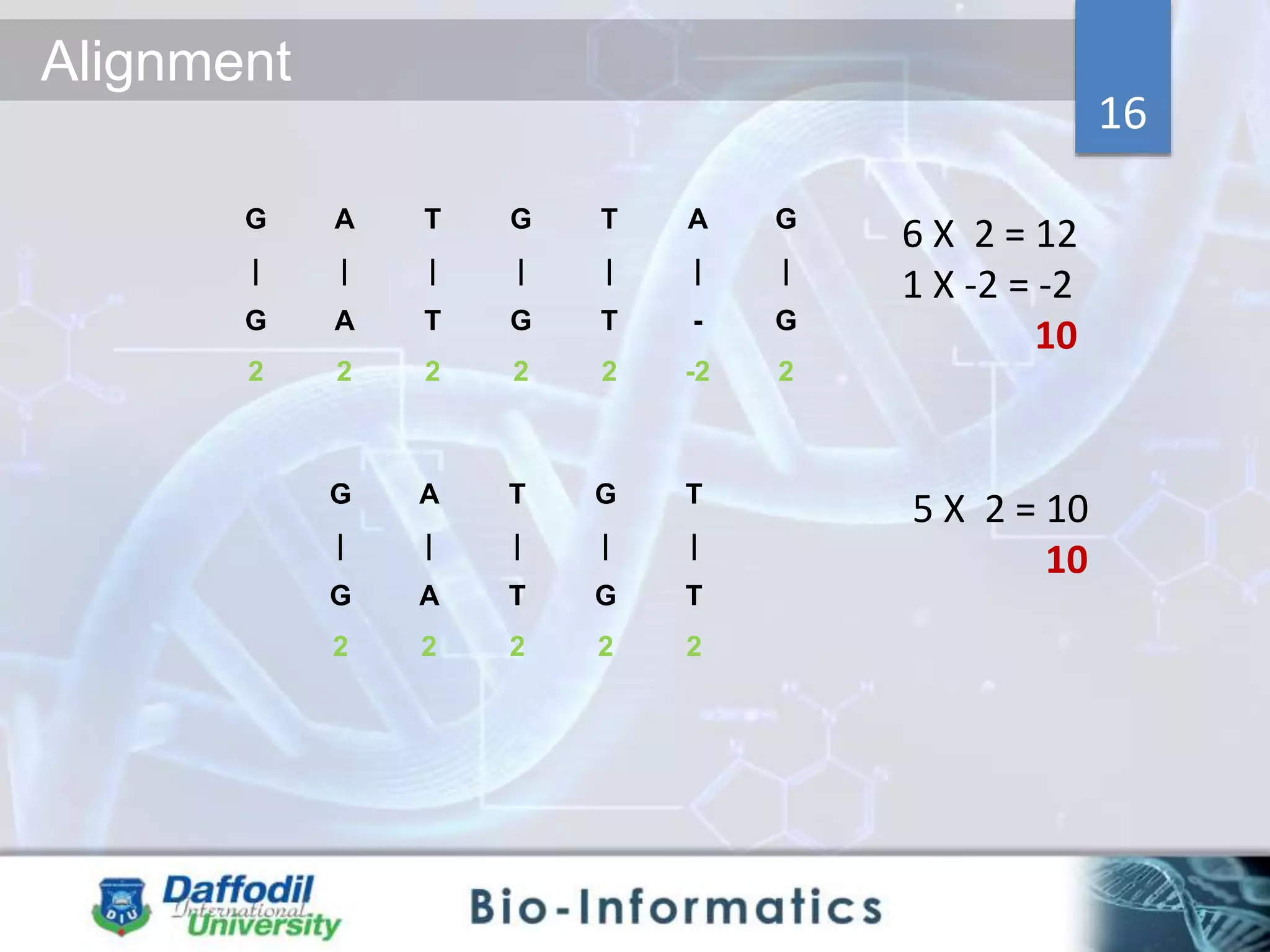
The document discusses sequence alignment, highlighting its importance in discovering functional, structural, and evolutionary relationships among genes and proteins. It explains the differences between global and local alignment techniques, specifically the Needleman-Wunsch algorithm for global alignment and the Smith-Waterman algorithm for local alignment. Detailed steps for scoring, matrix filling, and traceback processes for optimal alignment are provided.