Downloaded 192 times








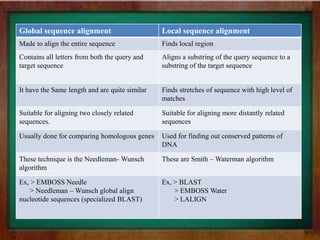







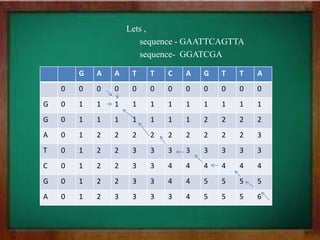





This document discusses global and local sequence alignment. Global alignment aims to align the entire sequences, treating gaps equally across the sequences. It is useful for closely related sequences of similar length. Local alignment finds locally similar regions, allowing gaps to be treated differently. It is useful for more distantly related sequences that may contain similar subsequences. Both use dynamic programming, with global alignment using Needleman-Wunsch and local using Smith-Waterman. Dynamic programming breaks the problem into subproblems by filling a matrix to find the highest scoring alignment.























































































