
















![Extreme Value Distribution Probability density function for the extreme value distribution resulting from parameter values = 0 and = 1, [ y = 1 – exp(- e -x )], where is the characteristic value and is the decay constant. y = 1 – exp(- e - ( x - ) )](https://image.slidesharecdn.com/sequencealignment-belgaum-110131042535-phpapp02/85/Sequence-alignment-belgaum-22-320.jpg)
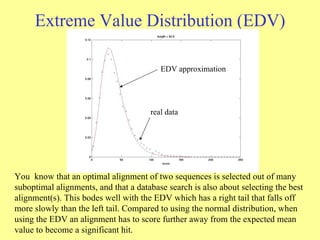




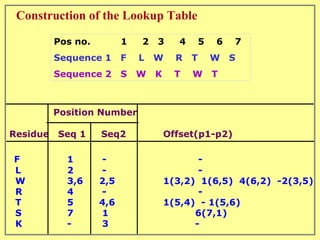
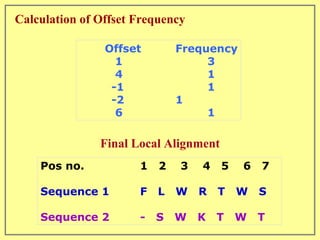







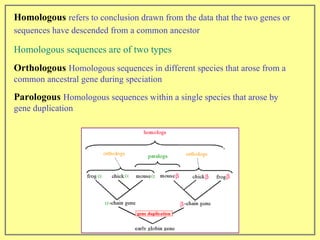
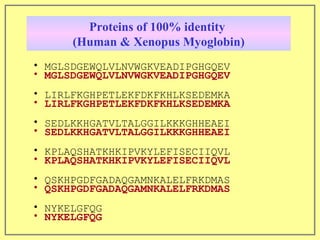
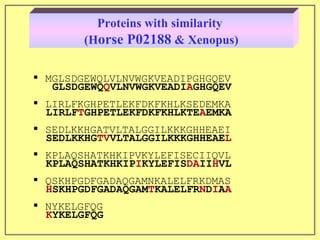
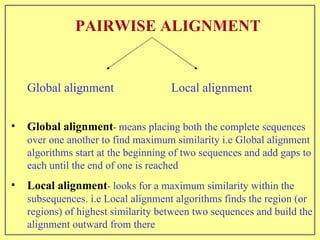
This document discusses sequence alignment, which involves placing two or more biological sequences in an optimal alignment to identify regions of similarity and deduce evolutionary relationships. It defines key terms like similarity, identity, conservation, and optimal alignment. It also describes the rationale for alignment, which is to compare sequences and find similarities that provide insights into biological function and evolutionary history. Finally, it outlines different types of alignment like global, local, pairwise, and multiple sequence alignment.























































































