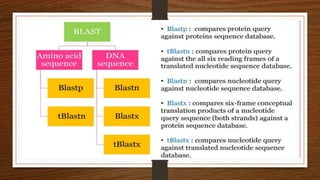
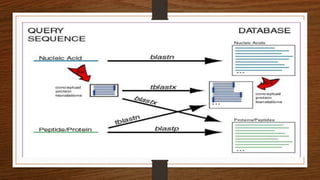
This document discusses global and local sequence alignment. It introduces sequence alignment and its uses in identifying similarities between sequences that could indicate functional or evolutionary relationships. It describes the principles of alignment and the different types of alignment, including global alignment, which aligns entire sequences, and local alignment, which matches regions of similarity. Methods for alignment include dot plots, scoring matrices, and dynamic programming. BLAST is introduced as a tool for comparing sequences against databases using local alignment algorithms.