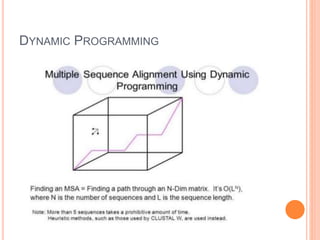
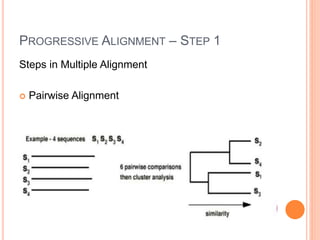
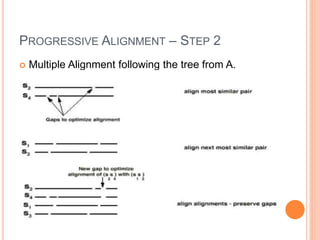
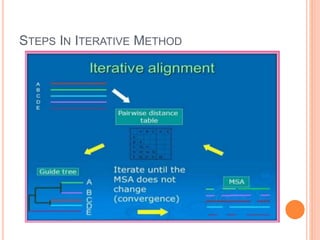
This document discusses multiple sequence alignment (MSA), which aligns three or more biological sequences, such as proteins or nucleic acids, that likely share evolutionary relationships. MSA can be used to infer homology and evolutionary relationships between sequences. There are different types of MSA, including dynamic programming, progressive, and iterative methods. Progressive methods first perform pairwise alignments and then align pairs of sequences together, while iterative methods improve on progressive methods by avoiding pairwise alignments. MSA has applications in characterizing protein families and detecting conserved motifs and structural similarities.