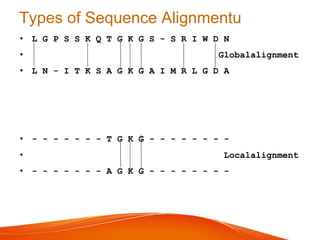
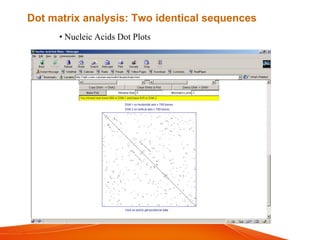
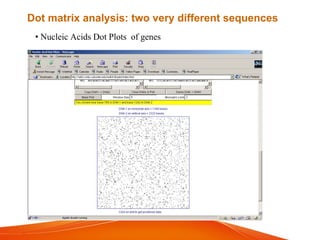
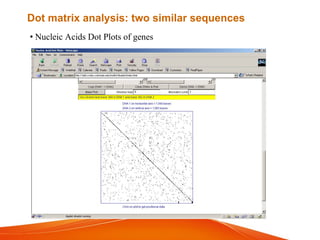
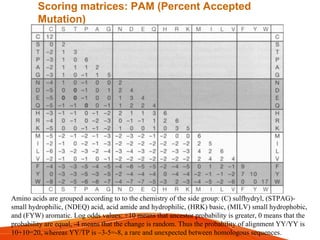
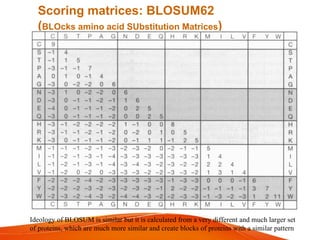
The document provides an overview of computational methods for sequence alignment. It discusses different types of sequence alignment including global and local alignment. It also describes various methods for sequence alignment, such as dot matrix analysis, dynamic programming algorithms (e.g. Needleman-Wunsch, Smith-Waterman), and word/k-tuple methods. Scoring matrices like PAM and BLOSUM that are used for sequence alignments are also explained.