Downloaded 11 times

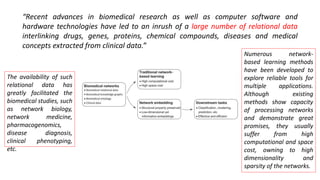


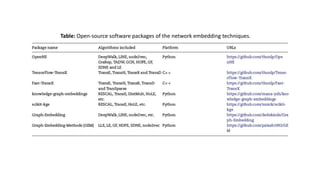
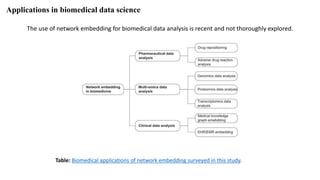













The document discusses the use of network embedding in biomedical data science, highlighting its potential to improve analysis of complex relational data concerning drugs, genes, and diseases. While network-based learning methods show promise, challenges such as high dimensionality, sparsity, and noise complicate their application. Opportunities for future development include combining local and global structural properties, dynamic embedding, and incorporating domain knowledge.





















































































