Downloaded 51 times






![● DLPC (1,2-dilauroyl-sn-glycero-3-phosphocholine) [12
carbon atoms]
● DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine)[14
carbon atoms]
● DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) [16
carbon atoms]
● DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine) [18
carbon atoms]](https://image.slidesharecdn.com/mcmdsofmembraneproteins-161019062537/85/Monte-Carlo-Simulations-Membrane-Simulation-and-Dynamics-8-320.jpg)
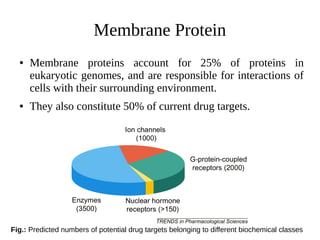












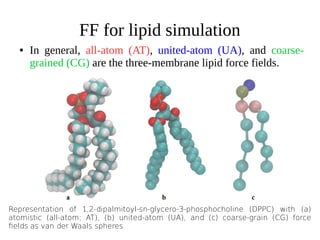







Monte Carlo simulations and molecular dynamics simulations are common computational methods to study membrane proteins and lipid bilayers. Monte Carlo simulations use random sampling to explore the behavior of complex systems. Molecular dynamics simulations numerically simulate particle motions under internal and external forces based on empirical energy functions. There are different levels of molecular dynamics simulations including atomistic, united atom, and coarse grained simulations, each with varying degrees of atomic detail and accessible timescales. Parameterized force fields are used to model interactions in lipid and protein systems. These computational methods provide insights into membrane and protein dynamics that are difficult to obtain experimentally.

































































































