Download as PDF, PPTX


![What’s next?
LDA uses local density ρ from homogenous
electron gas
Next step: Let’s add a gradient of the density!
Generalized gradient approximation (GGA)
NANO266
2
Exc
GGA
[ρ↑
,ρ↓
]= drρ(r)εxc (∫ ρ↑
,ρ↓
, ∇ρ↑
, ∇ρ↓
)](https://image.slidesharecdn.com/05-exchange-correlationfunctionals-150406231700-conversion-gate01/85/NANO266-Lecture-5-Exchange-Correlation-Functionals-2-320.jpg)

![Why stop at the first derivative?
Meta-GGA
Example: TPSS functional
NANO266
5
Exc
meta−GGA
[ρ↑
,ρ↓
]= drρ(r)εxc (∫ ρ↑
,ρ↓
, ∇ρ↑
, ∇ρ↓
,∇2
ρ↑
,∇2
ρ↓
)
Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Climbing the density functional ladder: nonempirical meta-generalized gradient
approximation designed for molecules and solids., Phys. Rev. Lett., 2003, 91, 146401, doi:10.1103/PhysRevLett.91.146401.](https://image.slidesharecdn.com/05-exchange-correlationfunctionals-150406231700-conversion-gate01/85/NANO266-Lecture-5-Exchange-Correlation-Functionals-5-320.jpg)



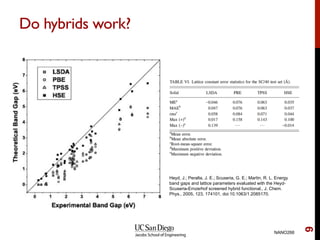
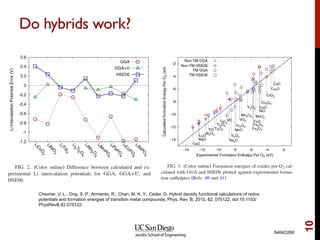


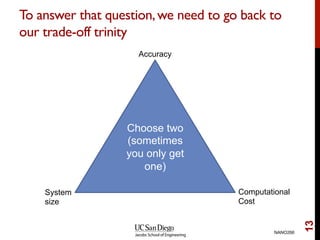
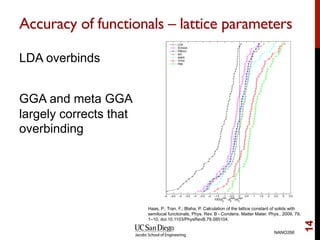
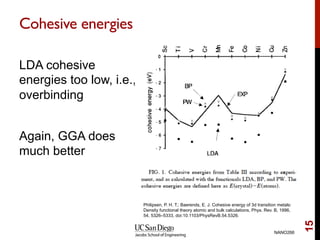



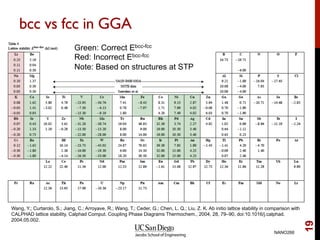
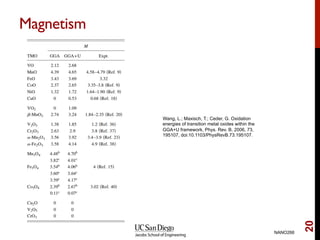
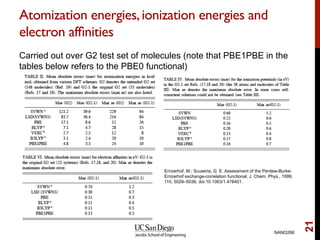
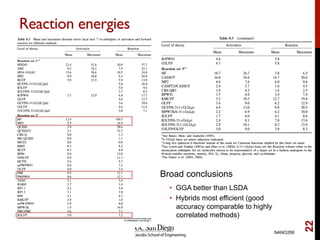
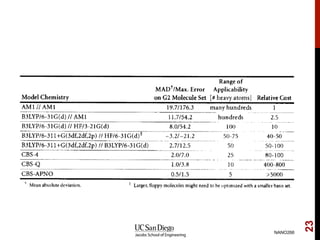


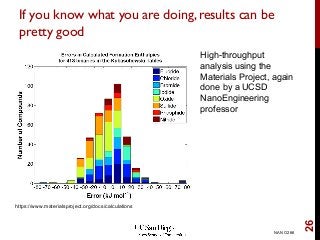

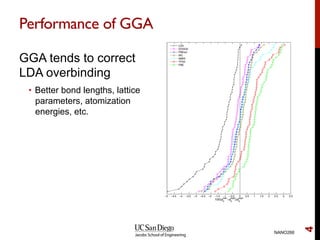
The document discusses advancements in exchange-correlation functionals used in Density Functional Theory (DFT), emphasizing the progression from Local Density Approximation (LDA) to Generalized Gradient Approximations (GGA) and meta-GGA functionals. It highlights specific functionals like BLYP, PBE, hybrid functionals like B3LYP, and their applications in improving accuracy for various properties like bond lengths, cohesive energies, and atomic structures. Additionally, it addresses the shortcomings of semi-local DFT, the need for hybrid functionals, and the accuracy trade-offs involved in DFT calculations.






































































































