Downloaded 43 times








![Electronic entropy
Probablity is given by Fermi-Dirac function
NANO266
12
hi
KS
= −
1
2
∇2
−
Zk
rik
+
k
∑
1
2
ρ(r')
ri − r'
dr∫∫ '+Vxc[ρ(r)]
hi
KS
ψi (r) =εiψi (r) Independent one-electron eigenstates can
be occupied or not
fi =
e−β(εi−εF )
1+e−β(εi−εF )
Sel = −kB fi ln( fi )+(1− fi )ln(1− fi )[ ]
i
∑](https://image.slidesharecdn.com/10-temperature-160504181430/85/NANO266-Lecture-10-Temperature-12-320.jpg)
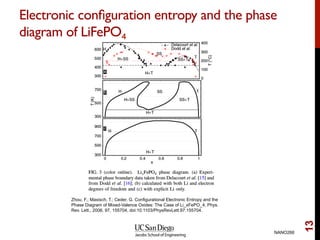



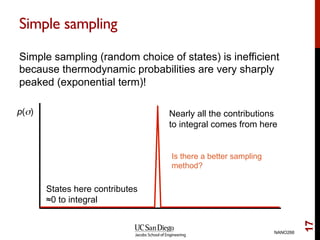


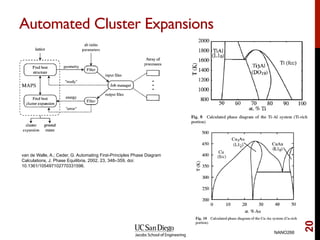
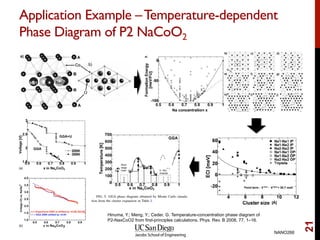


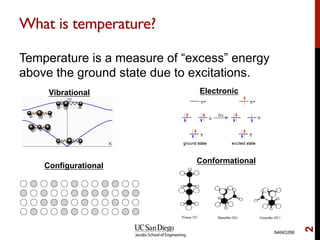
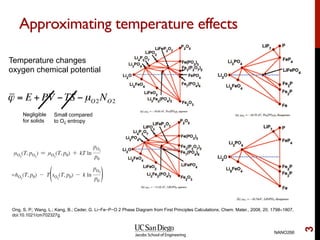
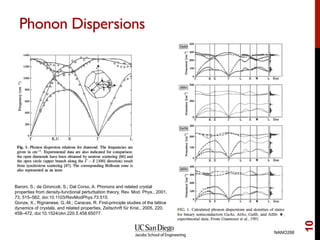
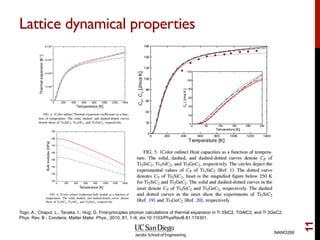
The document discusses the concept of temperature as a measure of energy above the ground state due to excitations, highlighting its effects on vibrational, configurational, and electronic properties in materials. Various methods for calculating acoustic properties such as phonons through density functional theory (DFT) and other approaches are examined, each with its advantages and disadvantages. It also touches on topics such as electronic entropy, phase diagrams, and methodologies for automating first-principles calculations for materials science.



















































































