Downloaded 166 times

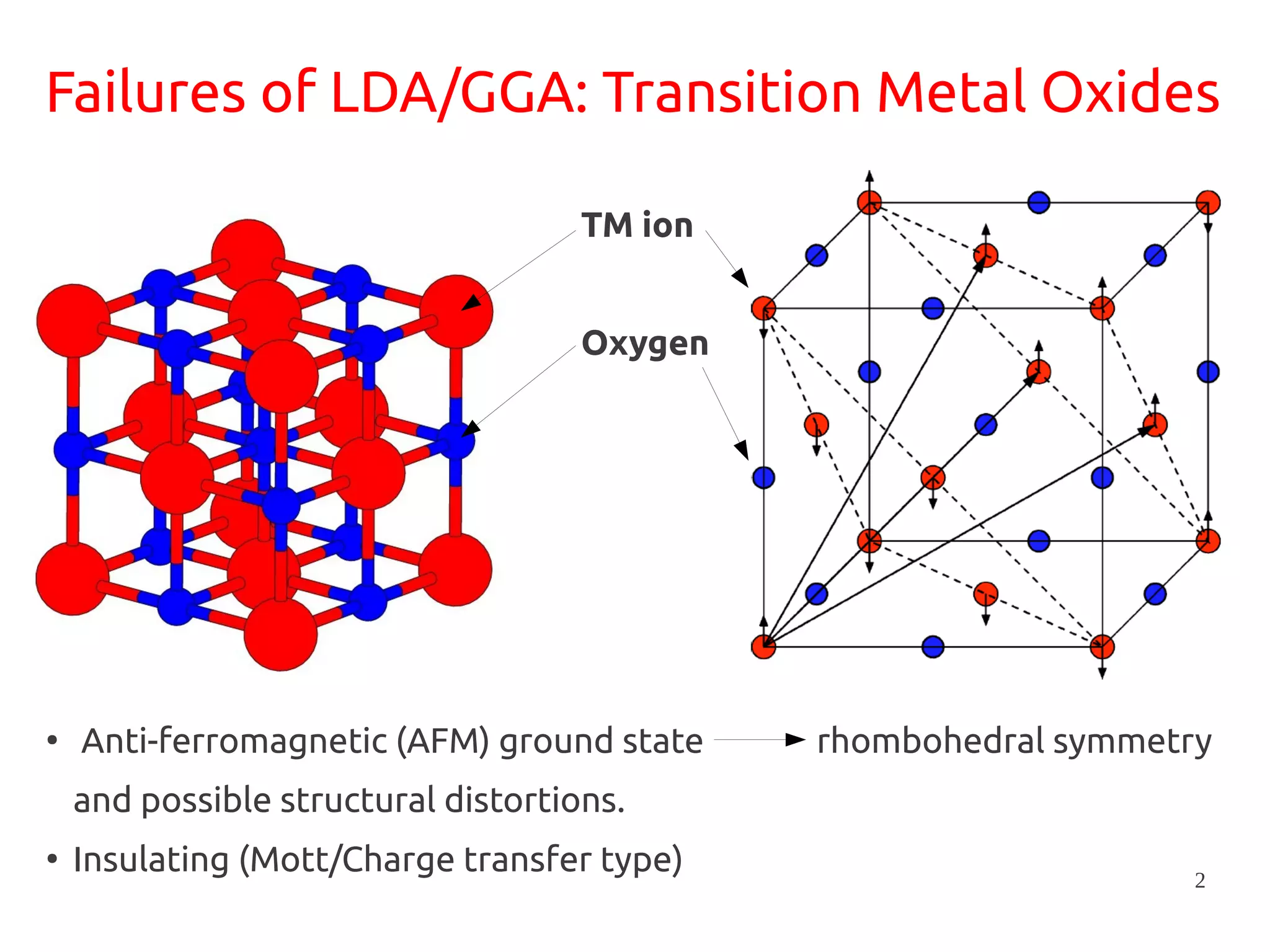
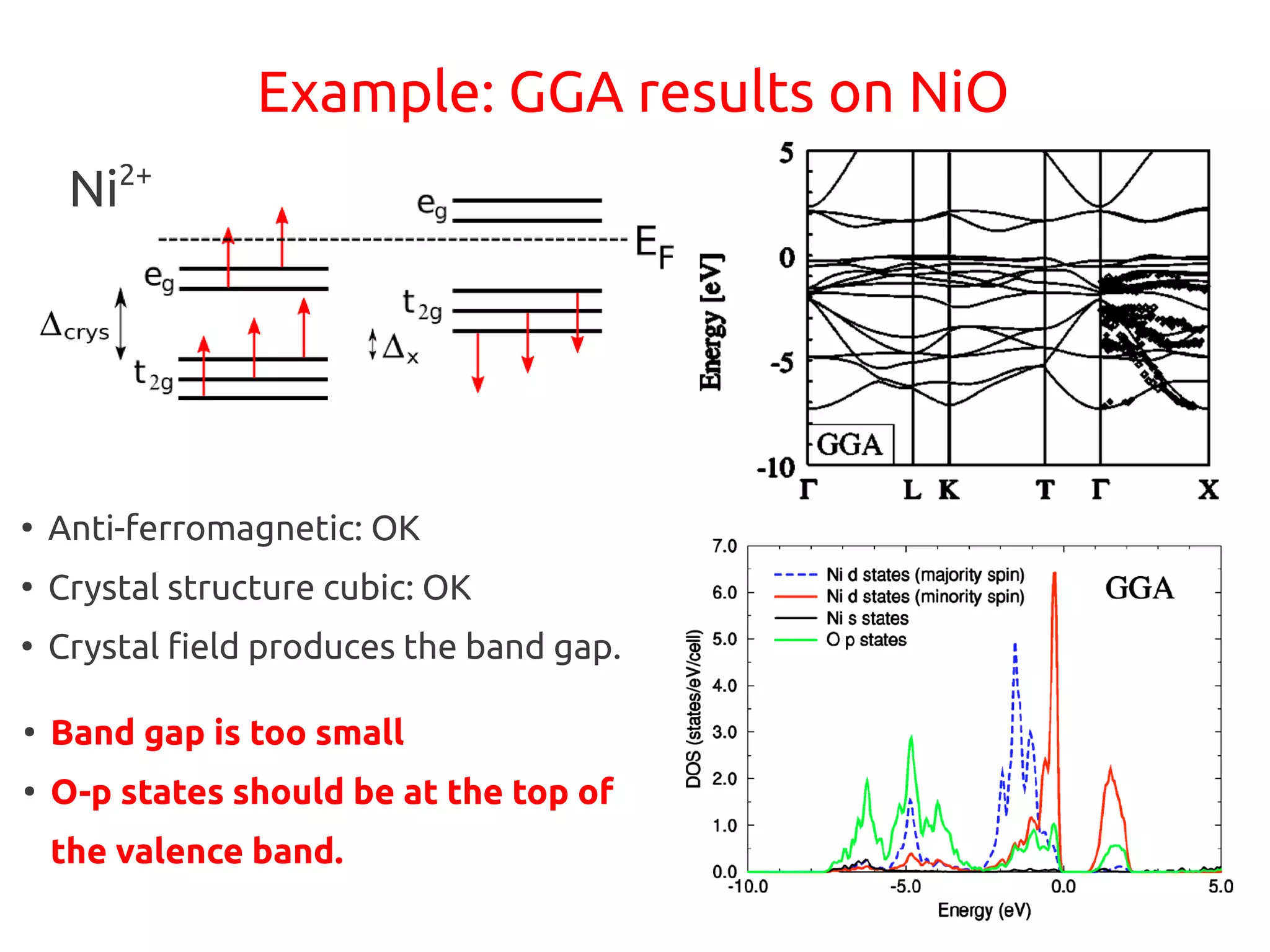
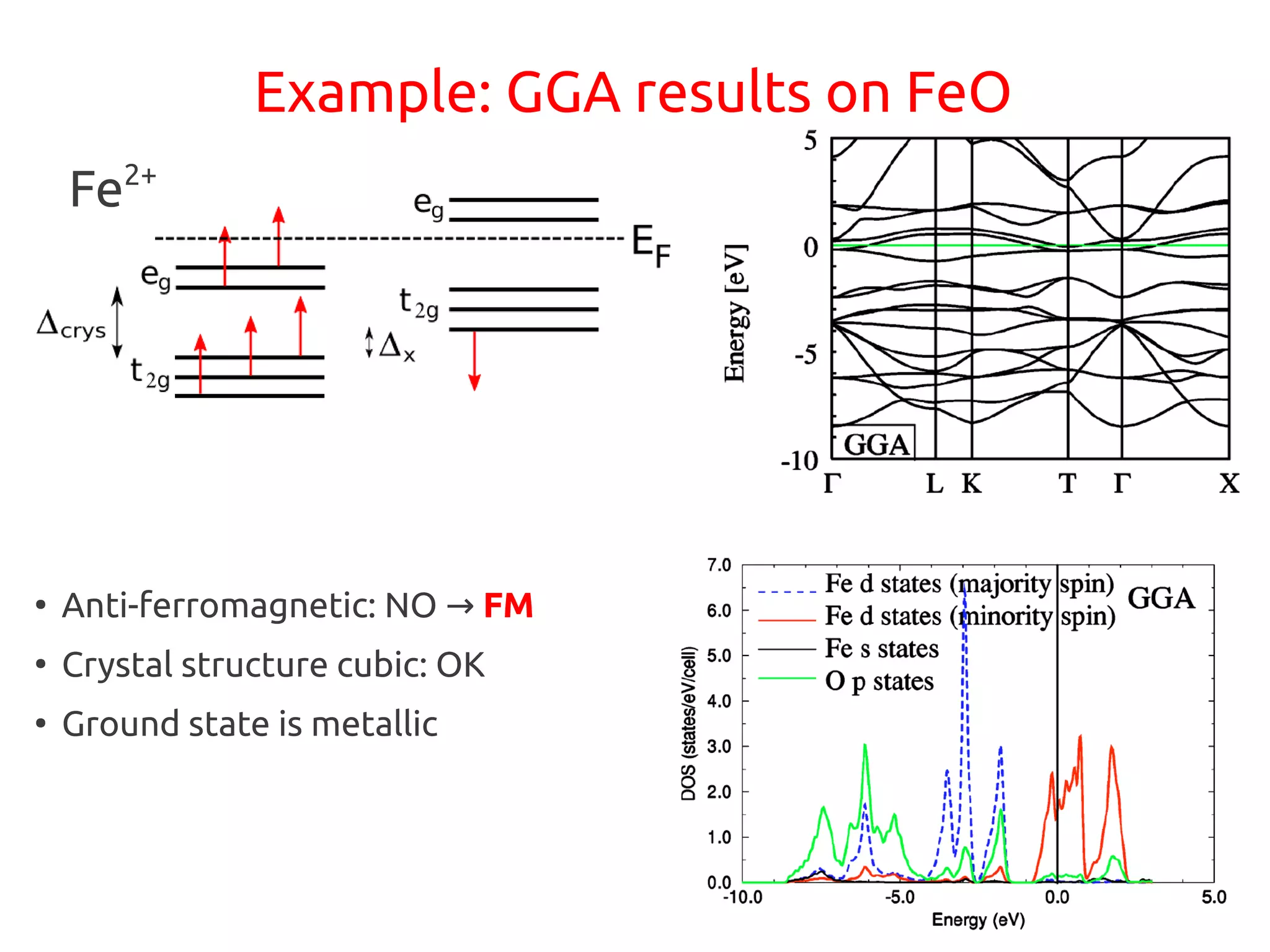
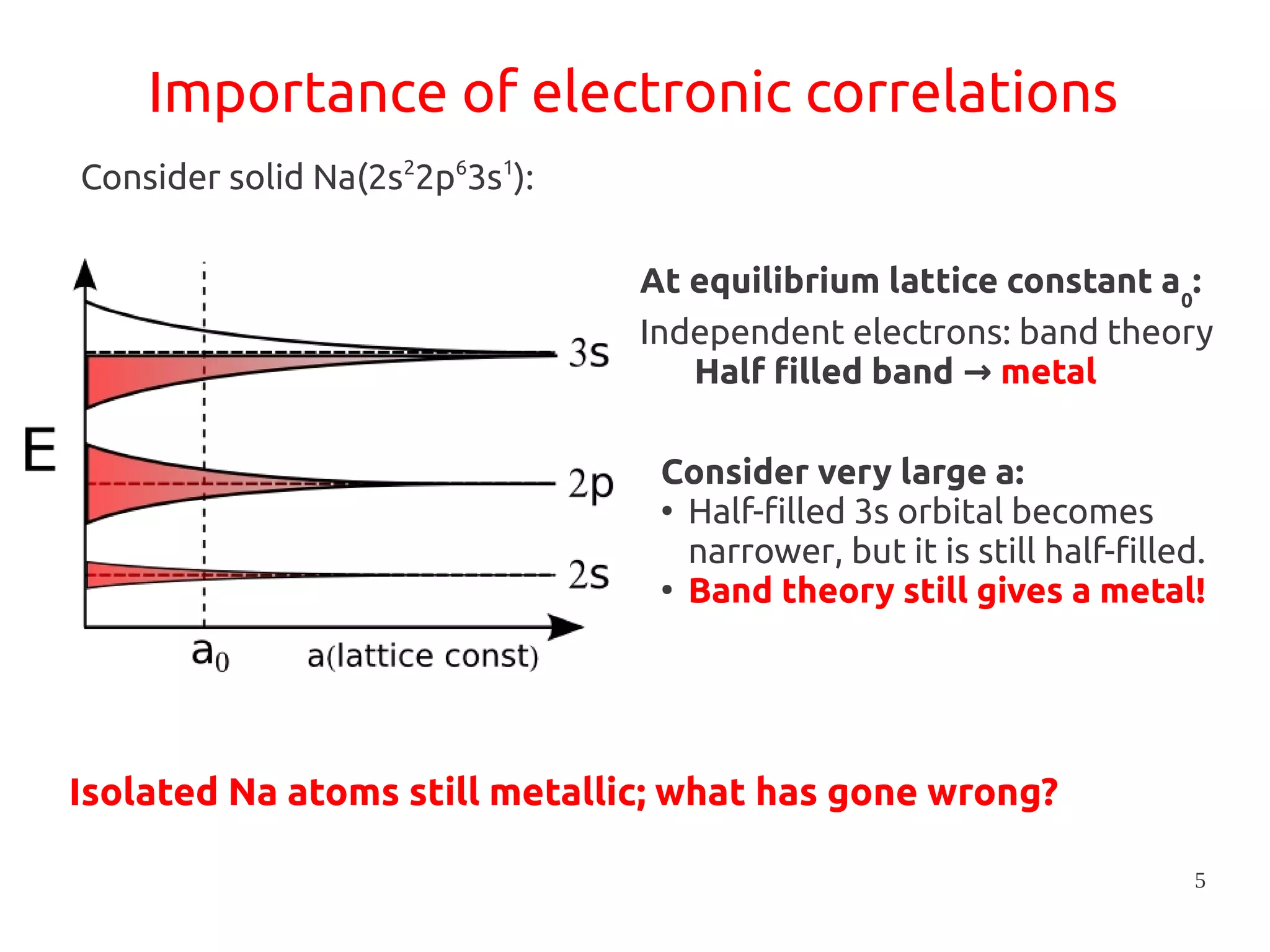



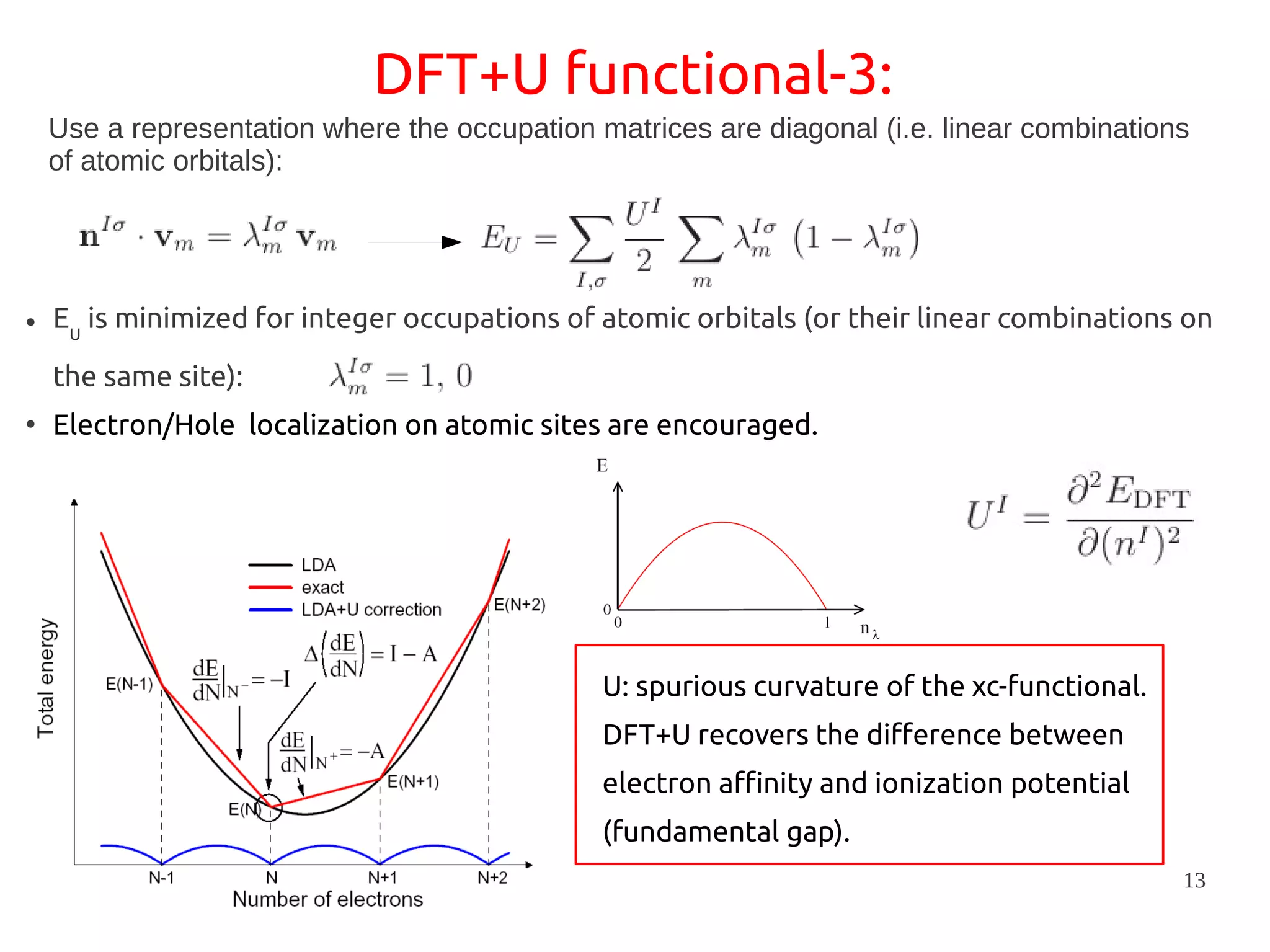
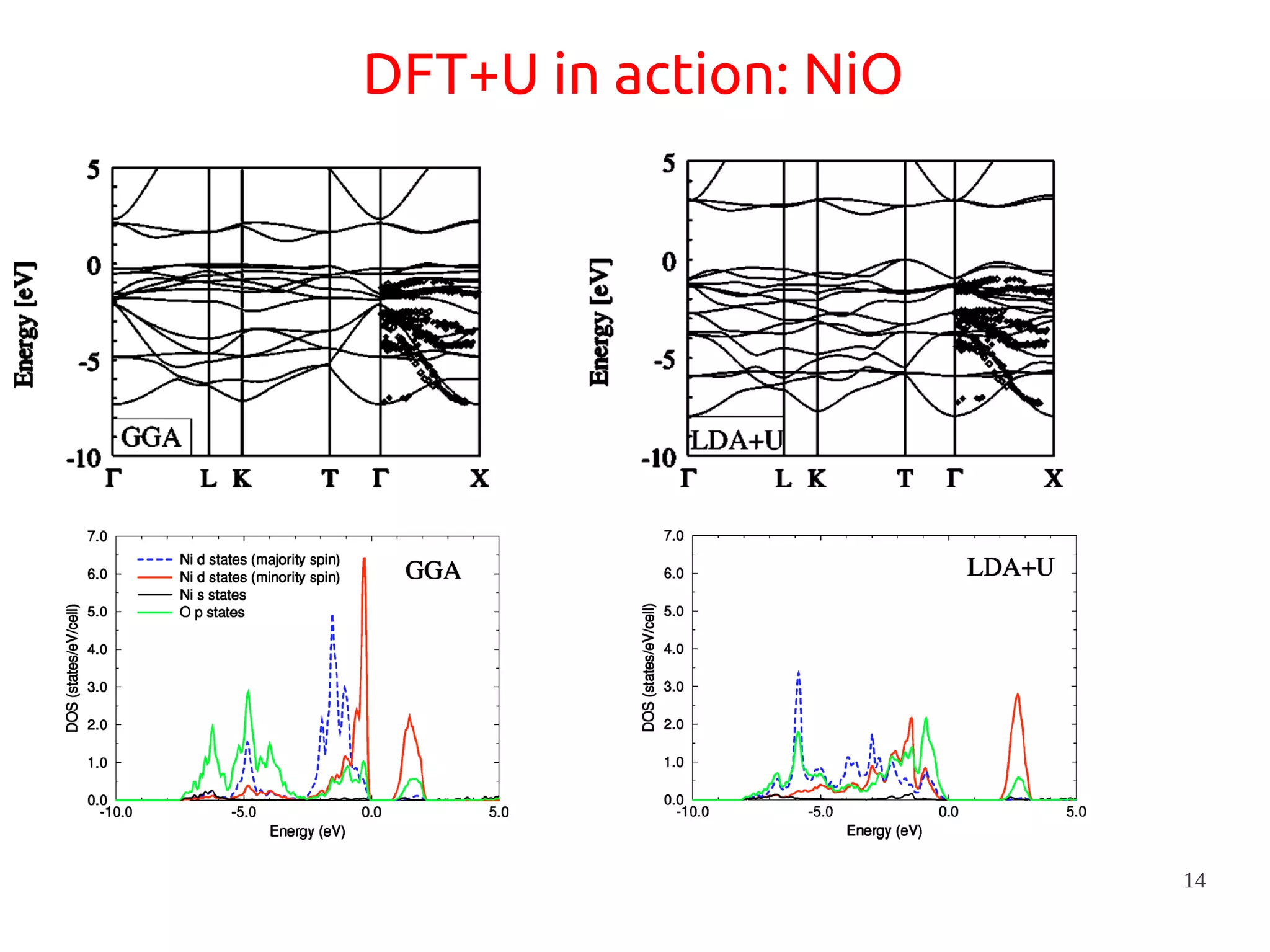
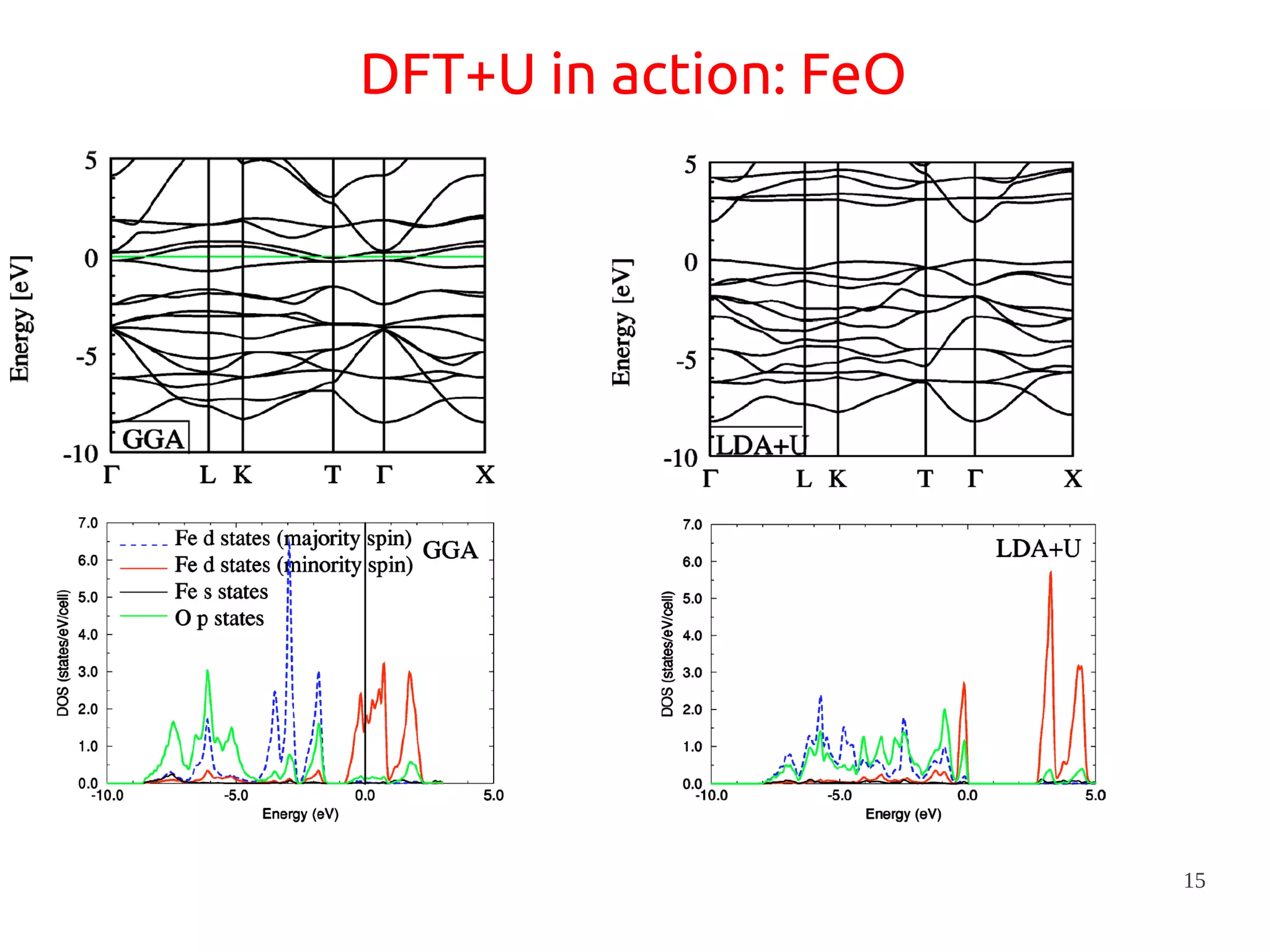


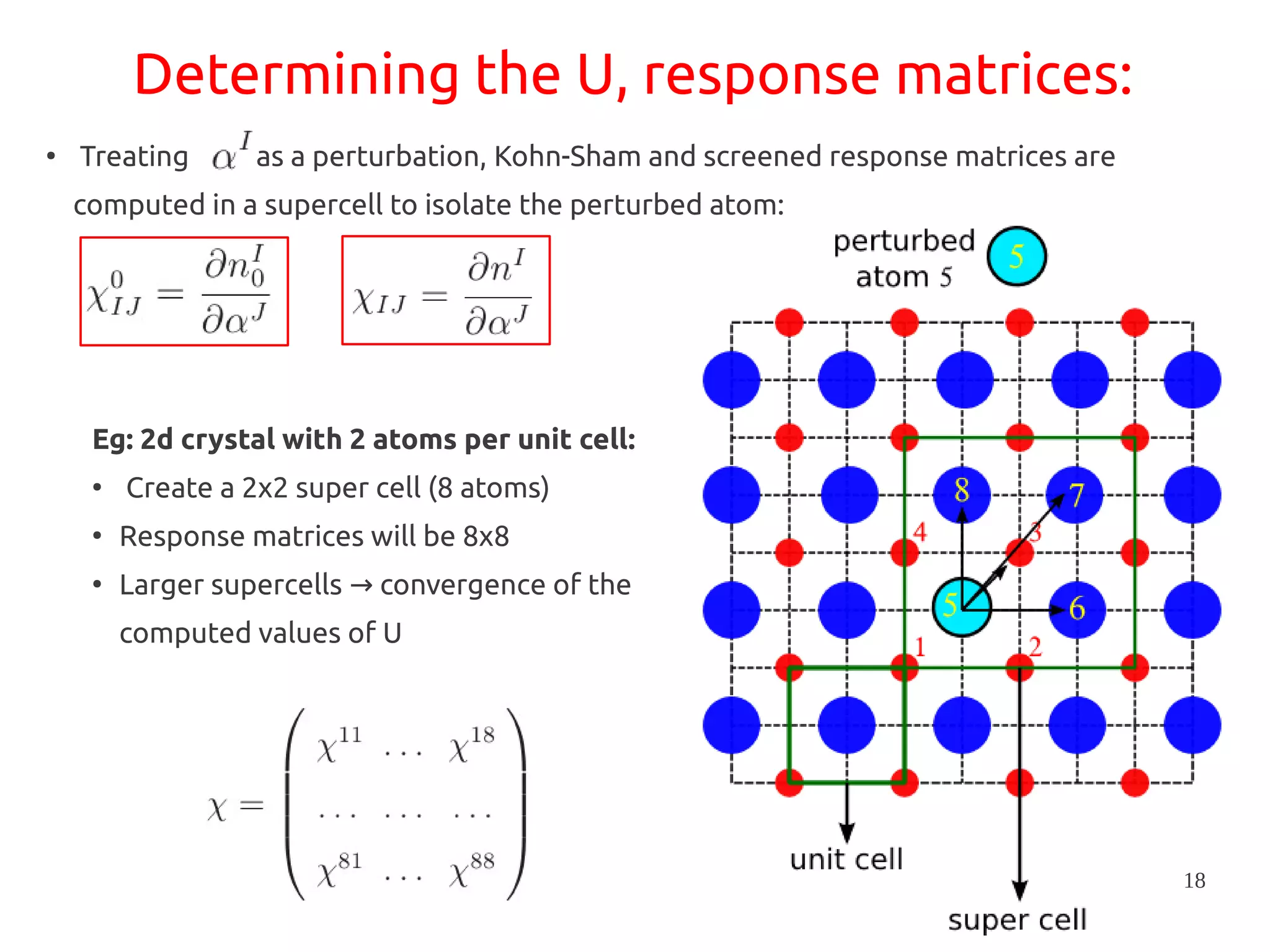
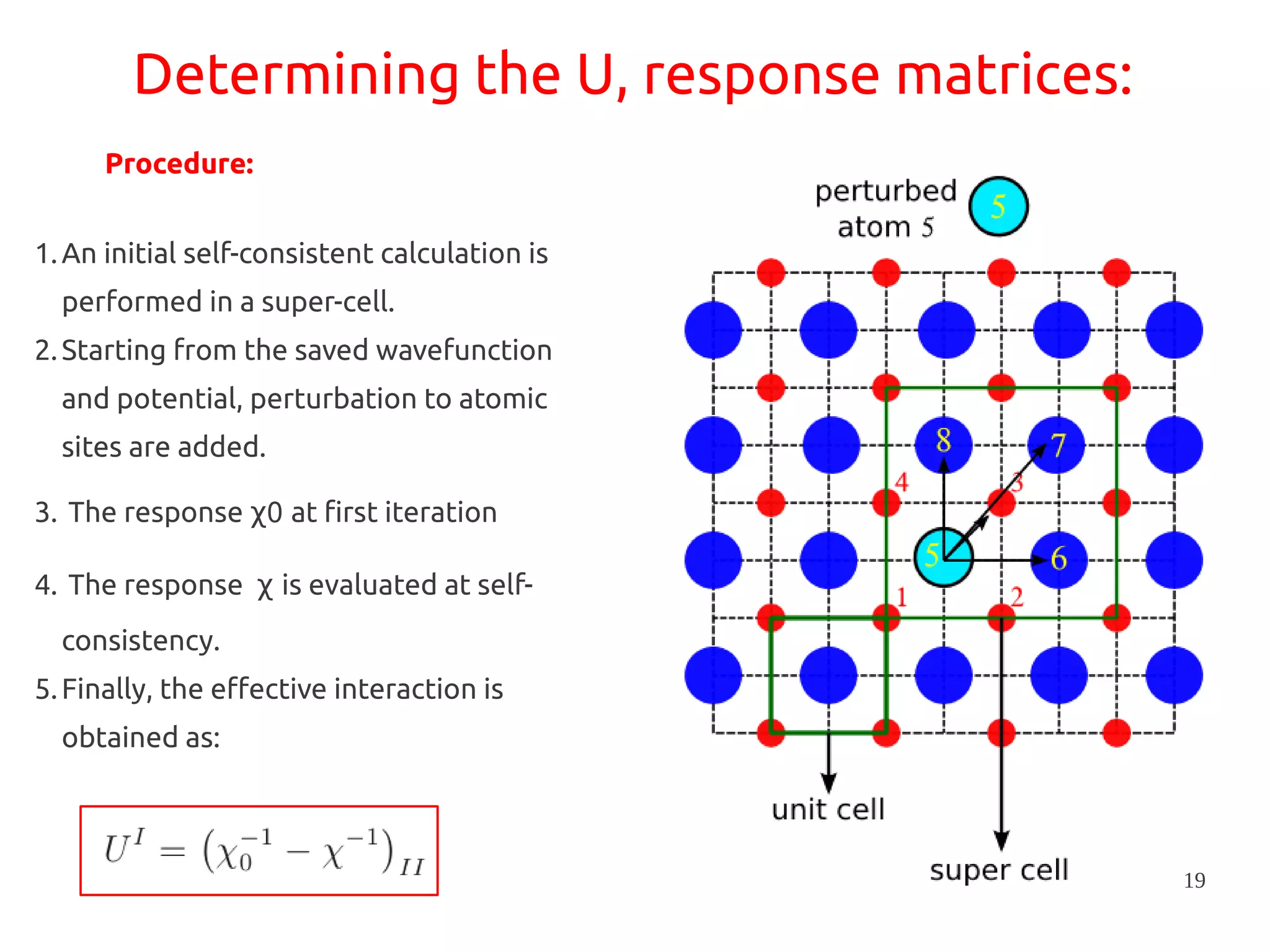
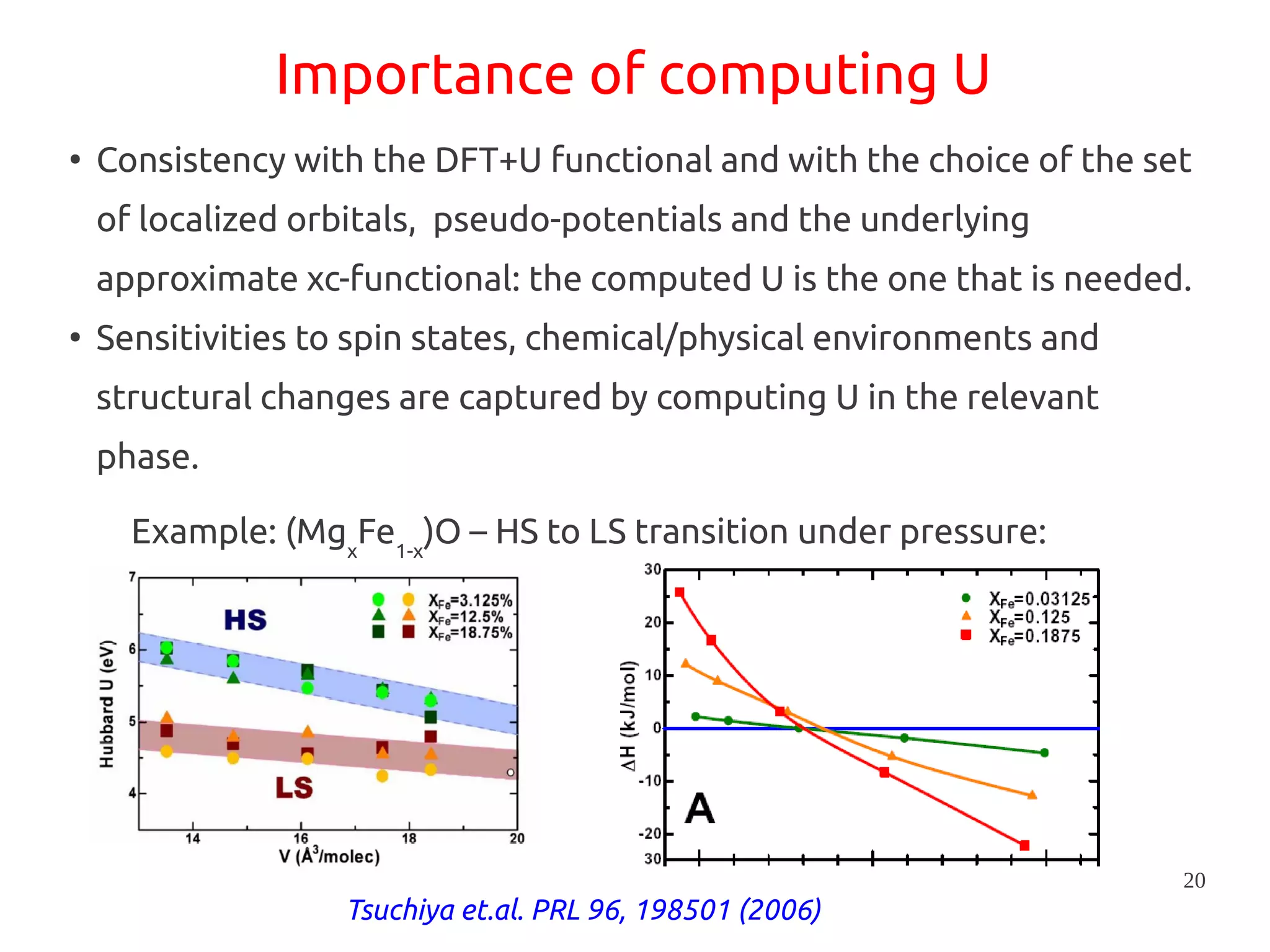
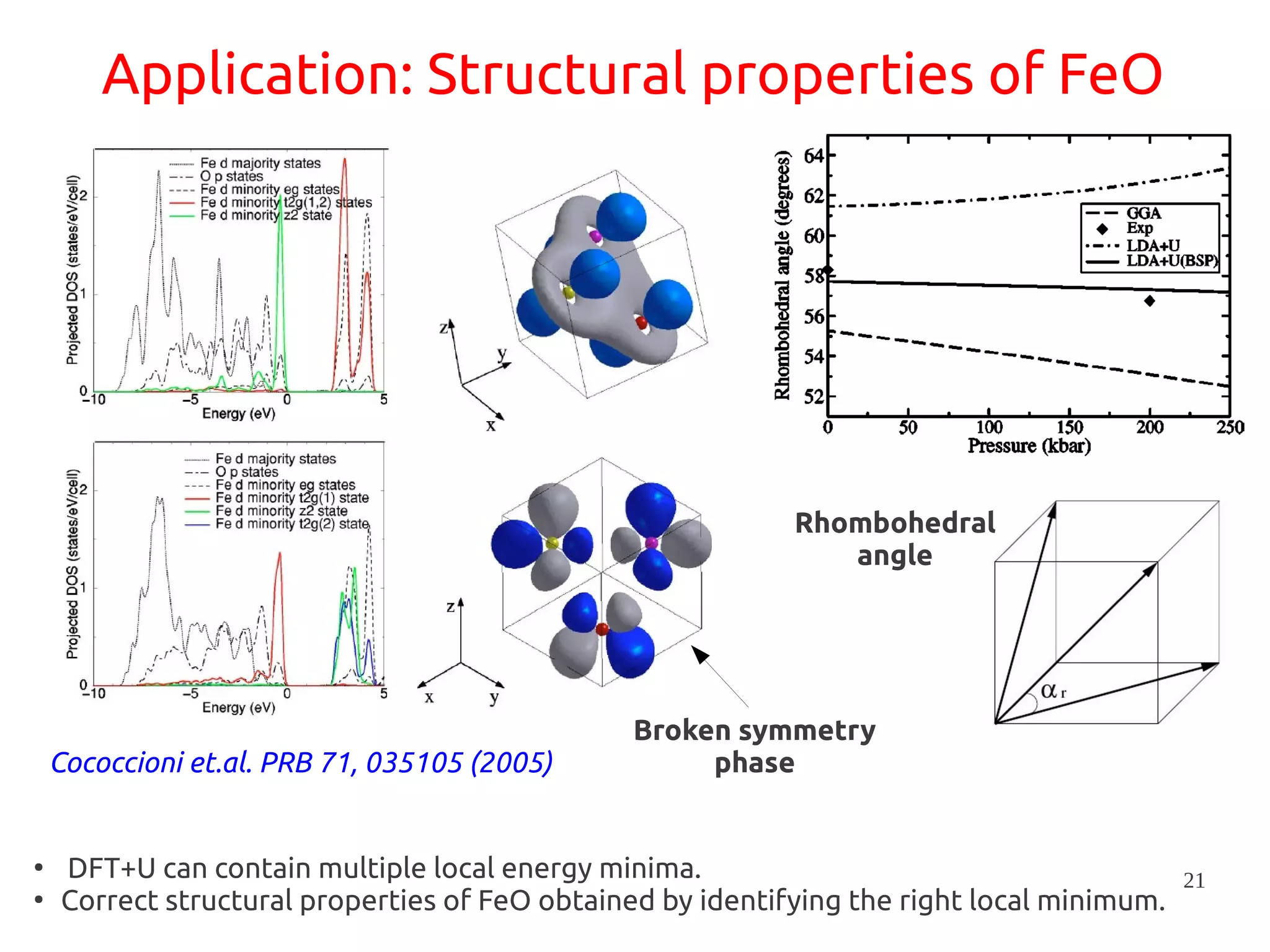
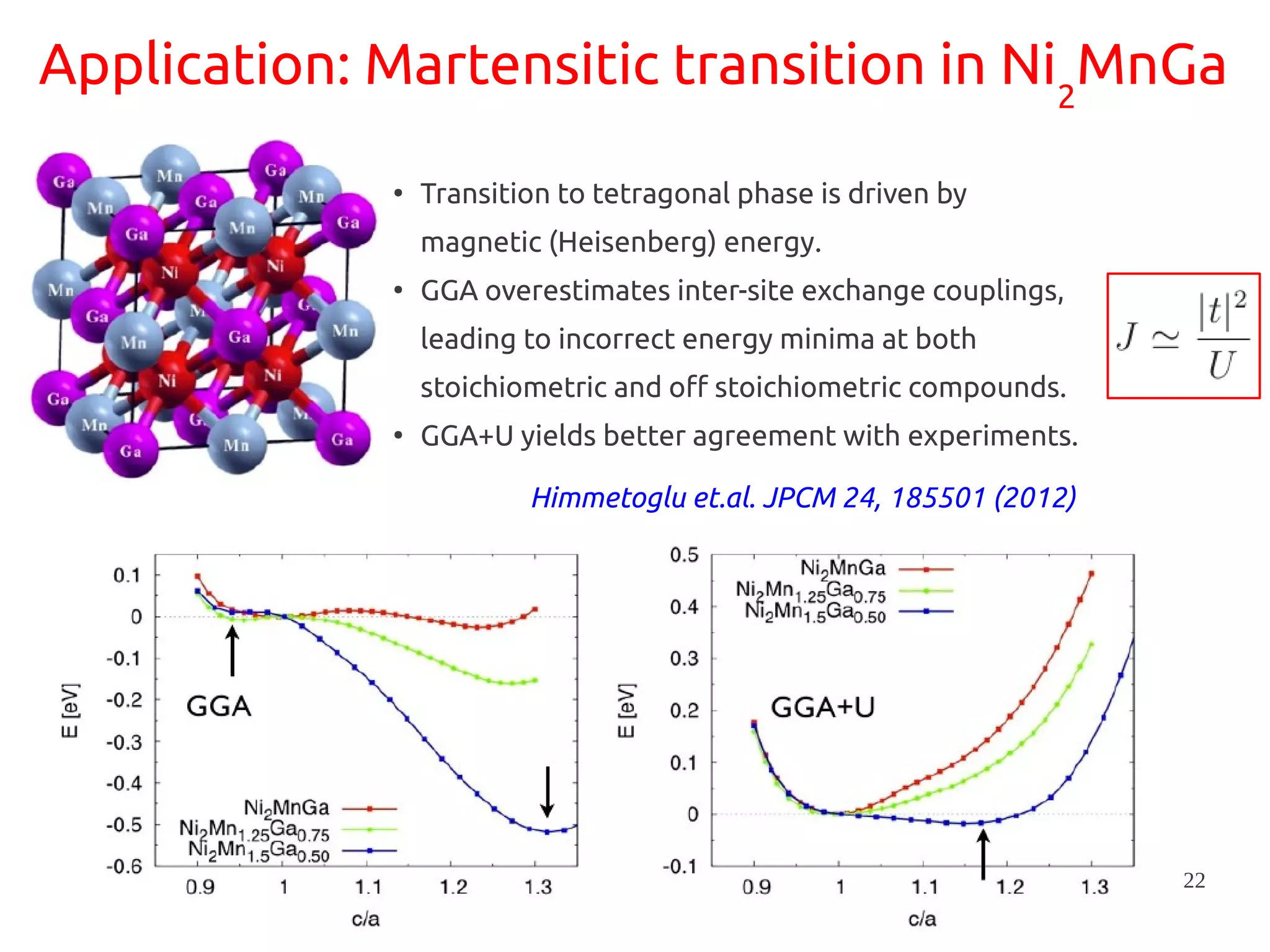
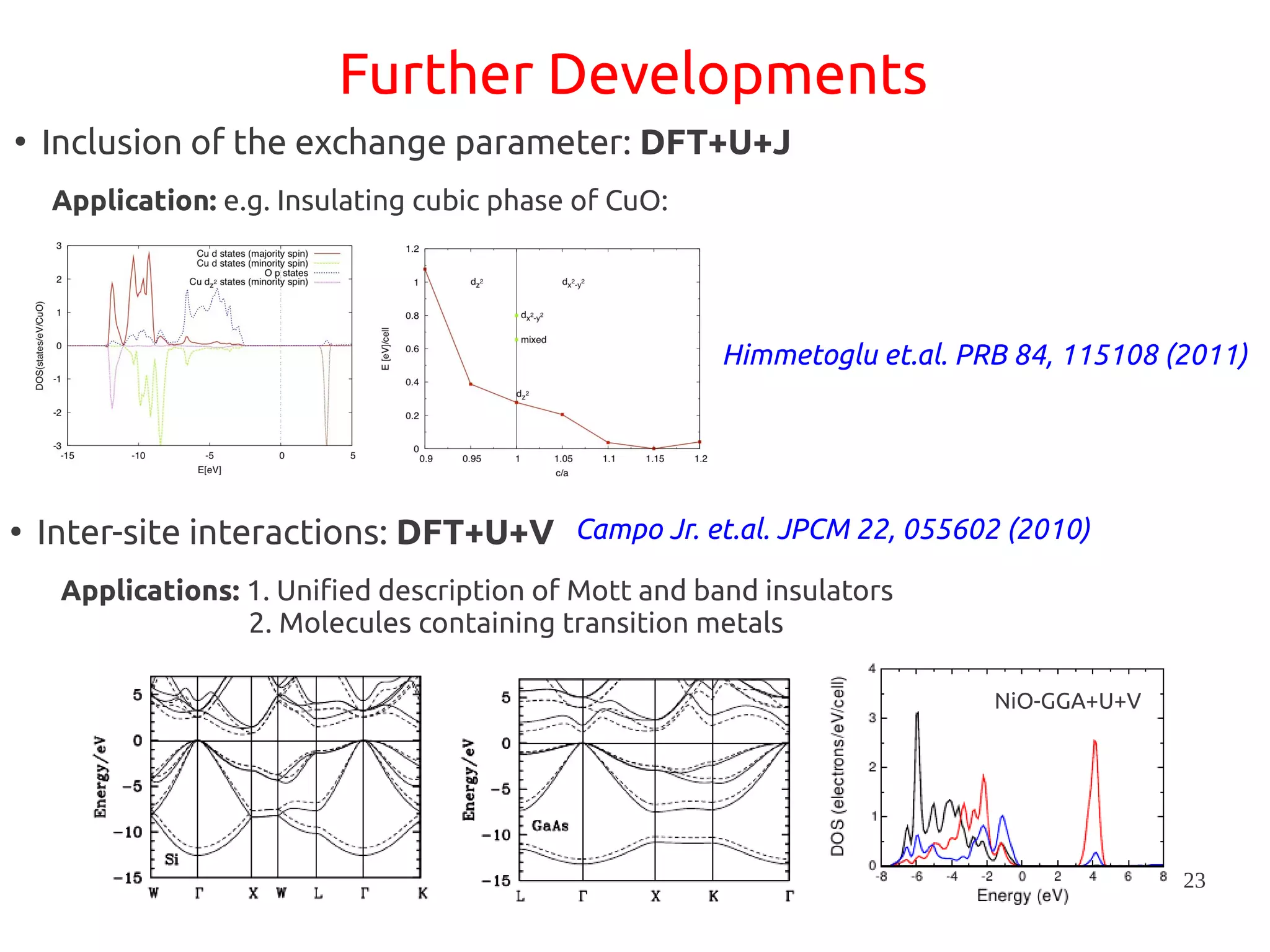

1. DFT+U is a method that adds Hubbard corrections to DFT to better account for localized electrons and electronic correlations in transition metal oxides that LDA/GGA cannot describe accurately. 2. It introduces an on-site Coulomb repulsion term U to the energy functional that favors electron localization and integer orbital occupations. 3. The U parameter can be computed using linear response theory by perturbing occupation matrices and evaluating screened response matrices in a supercell calculation.



































































