Downloaded 87 times


![What is molecular dynamics (MD)?
Moving atomic nuclei by solving Newton’s
equation of motion
For N nuclei, we have 3N positions and 3N
velocities, i.e., a 6N dimensional phase space.
NANO266
2
M!!RI = −
∂E
∂RI
= FI [{RJ}]](https://image.slidesharecdn.com/13-abinitiomoleculardyanmics-160504181814/85/NANO266-Lecture-13-Ab-initio-molecular-dyanmics-2-320.jpg)

![Initialization
2nd order PDF -> Initial positions and velocities
Positions
• Reasonable guess based on structure
• No overlap / short atomic distances
Velocities
• Small initial velocity
• Steadily increase temperature (velocity scaling)
NANO266
6
M!!RI = −
∂E
∂RI
= FI [{RI}]](https://image.slidesharecdn.com/13-abinitiomoleculardyanmics-160504181814/85/NANO266-Lecture-13-Ab-initio-molecular-dyanmics-6-320.jpg)

![Integrating equations of motion –Verlet algorithm
Key properties of Verlet algorithm
• Errors do not accumulate
• Energy is conserved
• Simulations are stable
NANO266
9
M!!RI = FI [{RJ}]
!!RI =
1
MI
FI [{RJ}]
[RI (t + Δt)− RI (t)]−[RI (t)− RI (t − Δt)]
(Δt)2
=
1
MI
FI [{RJ}]
RI (t + Δt) = 2RI (t)+ RI (t − Δt)+
(Δt)2
MI
FI [{RJ (t)}]](https://image.slidesharecdn.com/13-abinitiomoleculardyanmics-160504181814/85/NANO266-Lecture-13-Ab-initio-molecular-dyanmics-9-320.jpg)

![Coupled equations of motion
The CP Lagrangian
Equations of motion
NANO266
11
L =
1
2
(2µ)
i=1
N
∑ dr !ψi (r)
2
∫ + MI
I=1
NI
∑ !!RI − E[ψi,r]+ Λij drψi
*
(r)ψj (r)−δij∫%
&
'
(
ij
∑
Imposes orthonormality of
electronic states
µ !!ψi (r,t) = −Hψj (r)+ Λikψk (r,t)
k
∑
MI
!!RI = −
∂E
∂RI](https://image.slidesharecdn.com/13-abinitiomoleculardyanmics-160504181814/85/NANO266-Lecture-13-Ab-initio-molecular-dyanmics-11-320.jpg)





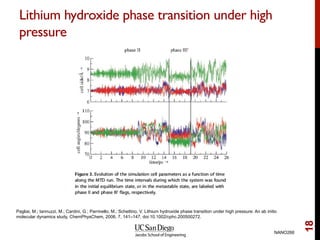
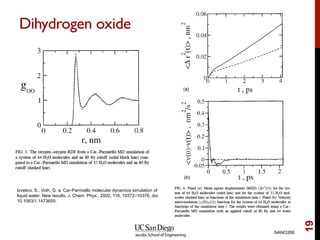

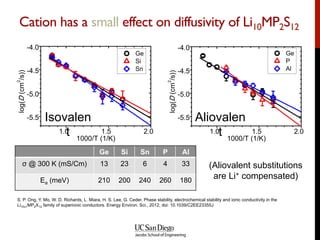



The document discusses the principles and applications of ab initio molecular dynamics (MD), focusing on computational methods for simulating atomic and molecular systems. It outlines the challenges of force calculations, various potential models, integration techniques, and different forms of quantum MD, such as Born-Oppenheimer and Car-Parrinello. Additionally, it highlights the uses of MD in studying thermodynamics, reaction dynamics, and material properties across diverse systems.






































































































