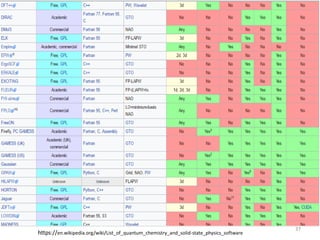
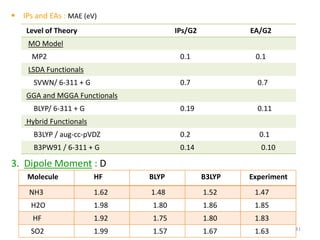
Density functional theory (DFT) provides an alternative approach to calculate properties of molecules by working with electron density rather than wave functions. DFT relies on two theorems linking the ground state energy and electron density. Approximations must be made for the exchange-correlation functional, with popular approximations including LDA, GGA, and hybrid functionals. DFT calculations can determine properties like molecular geometries, energies, vibrational frequencies, and more using software packages. While computationally efficient, DFT has limitations such as its reliance on approximate exchange-correlation functionals.


![INTRODUCTION
In the period 1995-2000, density functional theory (DFT) showed
a meteoric rise to popularity in quantum-chemistry
calculations:
“a substantial majority of the [quantum chemistry] papers
published today are based on applications of the density
functional theory” [K. Raghavachari, Theor. Chem. Acc., 103,
361 (2000)].
In DFT, one does not attempt to calculate the molecular wave
function. Instead, one works with the electron probability
density, ρ(x,y,z).
Its advantages include less demanding computational effort, less
computer time, and in some cases better agreement with the
experimental values than is obtained from Hartree-Fock
procedures.
3](https://image.slidesharecdn.com/dft-presentation-170319063157/85/Dft-presentation-3-320.jpg)








![DENSITY FUNCTIONAL THEORY
-FROM WAVE FUNCTION TO ELECTRON DENSITY
• Hohenberg and Kohn- at the heart of DFT(1964)
THEOREM 1: The ground state energy E is a
unique functional of electron density:
E = Eo[ρo(r)]
where ρ (r) represents the density function
which itself is a function of position (r).
12](https://image.slidesharecdn.com/dft-presentation-170319063157/85/Dft-presentation-12-320.jpg)
![DENSITY FUNCTIONAL THEORY
-FROM WAVE FUNCTION TO ELECTRON DENSITY
THEOREM 2: The electron density that
minimizes the energy of the overall functional
is the true ground state electron density:
E[ρ(r)]>Eo[ρo(r)]
13](https://image.slidesharecdn.com/dft-presentation-170319063157/85/Dft-presentation-13-320.jpg)

![EXCHANGE-CORRELATION
FUNCTIONAL, Exc[n(r)]
• Includes all quantum mechanical terms.
• Not known-needs to be approximated.
• It is a smaller fraction of the total energy.
• Improved results are obtained by relating
exchange correlation with the first derivative
of density.
• Both exchange-correlation are long and short
distance.
• Long distance exchange-correlation is static
and short distance is dynamic. 15](https://image.slidesharecdn.com/dft-presentation-170319063157/85/Dft-presentation-15-320.jpg)





![H{H-F} =T(K.E) + V(e-e) + V(N-e) + E(Exchange)
H{K-S} =[T(K.E,Ks) +V(e-e,ks) +V(N-e)]+{T(K.E) -T(K.E,Ks)}+
{V(e-e) - V(e-e,Ks)} + Ex
=[T(K.E,Ks) + V(e-e,ks) + V(N-e)]+ [∆T + ∆V + Ex]
=T(K.E,Ks) + V(e-e,ks) + V(N-e) + Vxc
21](https://image.slidesharecdn.com/dft-presentation-170319063157/85/Dft-presentation-21-320.jpg)






![Generalized Gradient
Approximation(GGA)
• Why do we need gradient corrected functional?
Ex(BECK)
• Becke’s exchange functional is (commonly used Ex [GGA])
= Ex (LSDA)+ ∆ Ex (BECK)
• Commonly used GGA Ec[GGA] include the Lee–Yang–Parr
(LYP) functional
28](https://image.slidesharecdn.com/dft-presentation-170319063157/85/Dft-presentation-28-320.jpg)