Download as PDF, PPTX




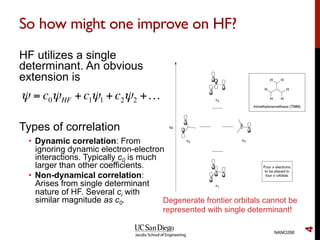

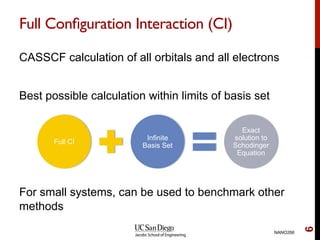






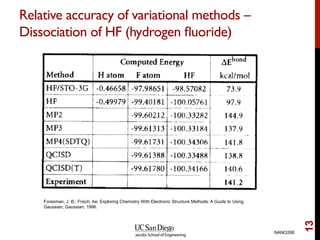
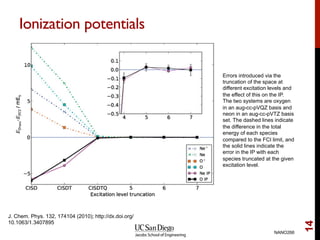
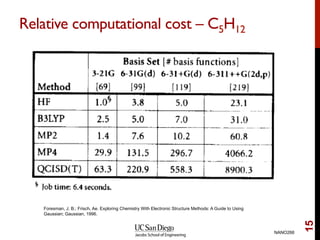




The document discusses the limitations of the Hartree-Fock (HF) approximation in quantum chemistry, particularly regarding the treatment of electron correlation. It explores various advanced methods, such as Møller-Plesset perturbation theory and coupled-cluster methods, for improving upon HF, emphasizing the importance of understanding both dynamic and non-dynamical correlation. Additionally, it highlights the computational costs associated with these methods and provides references for further detailed study.























































































