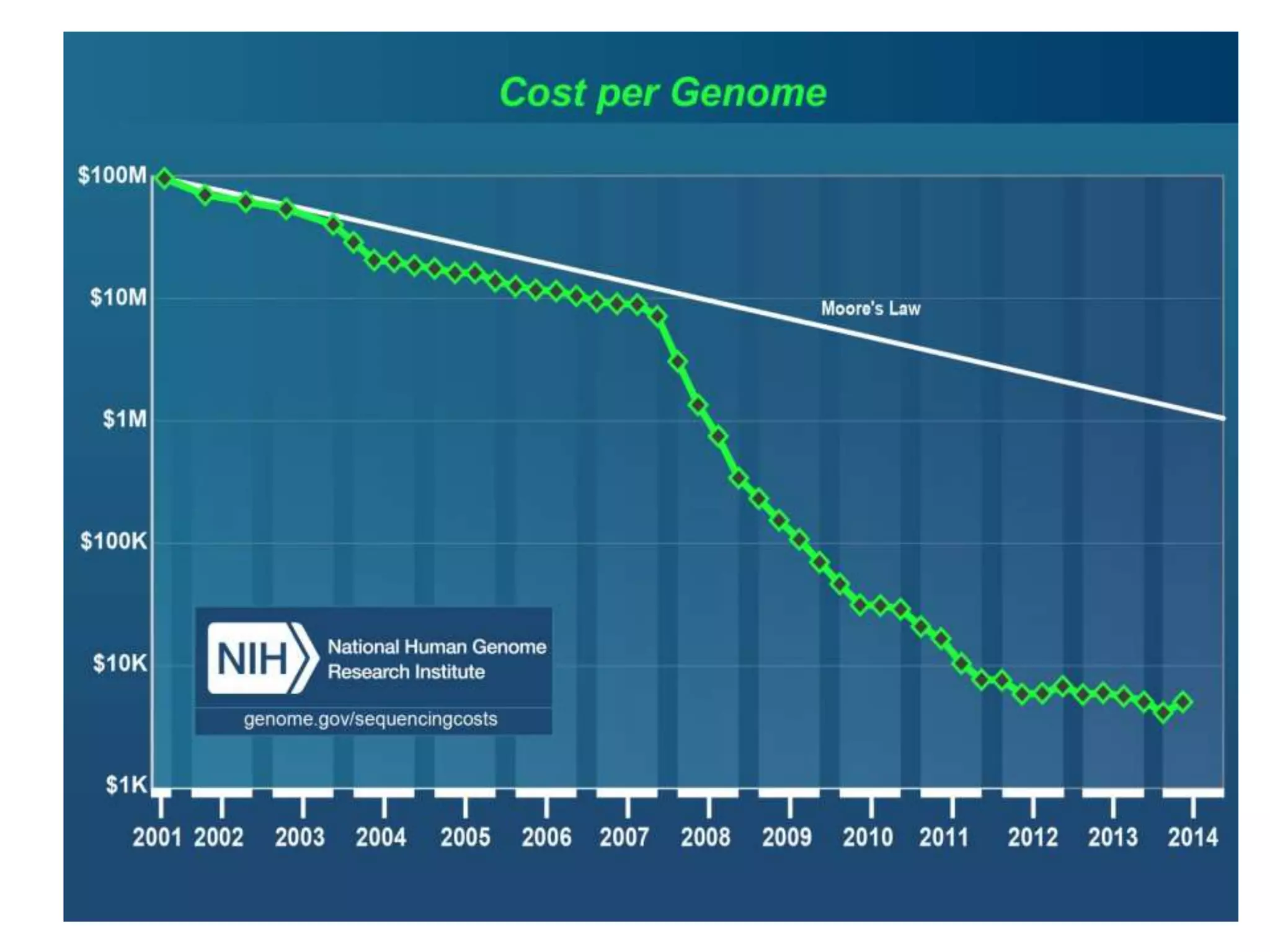
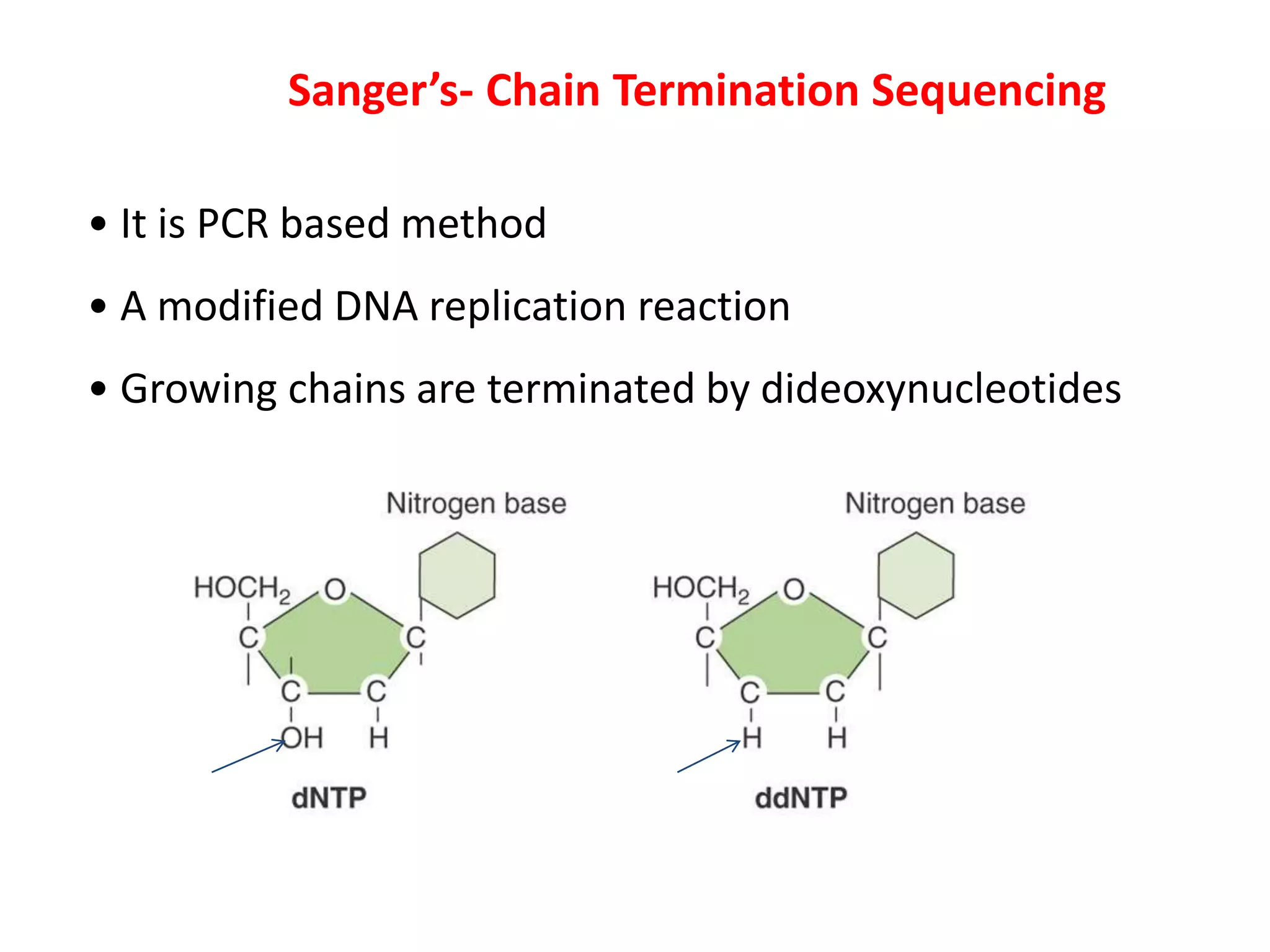
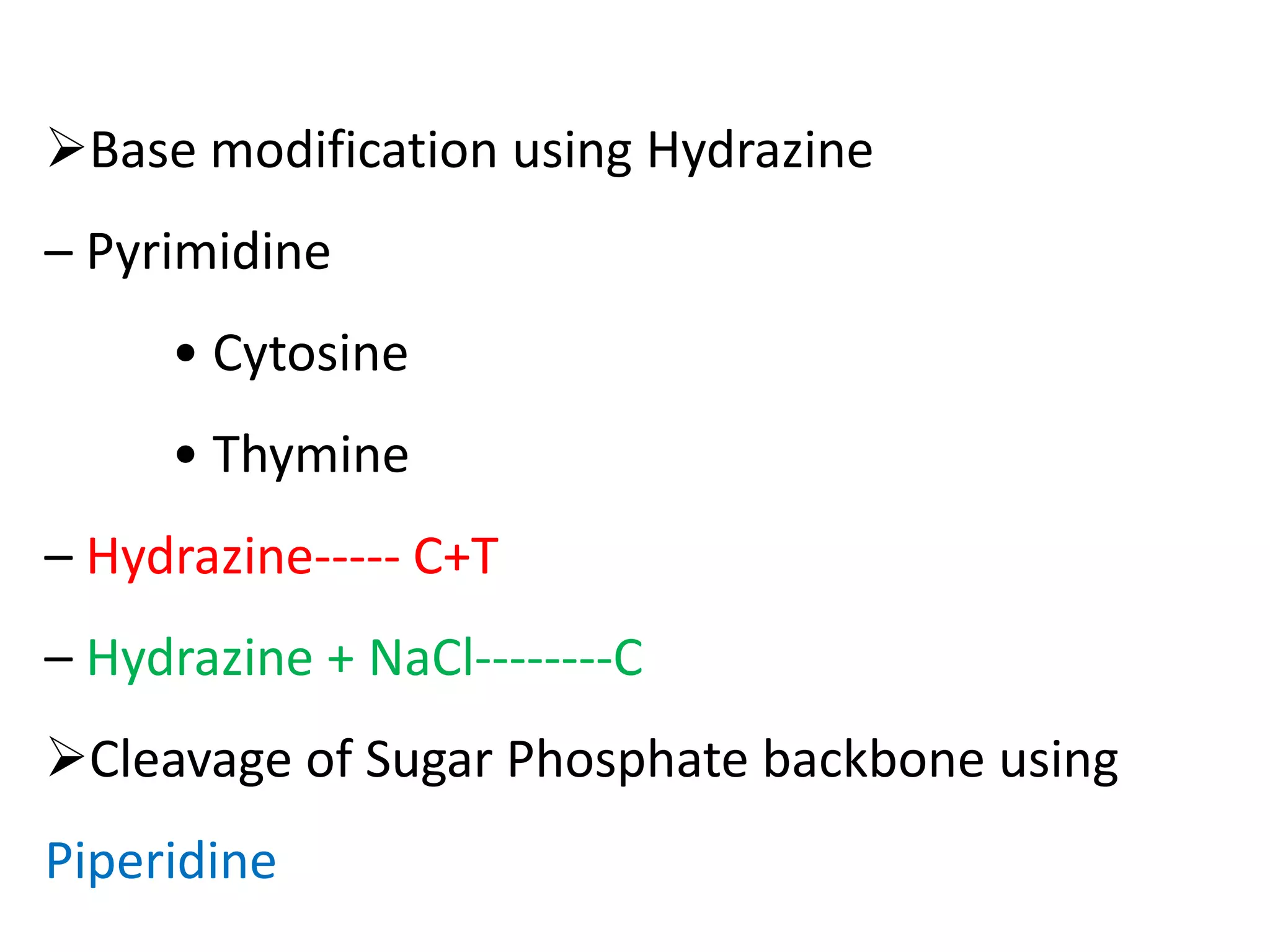
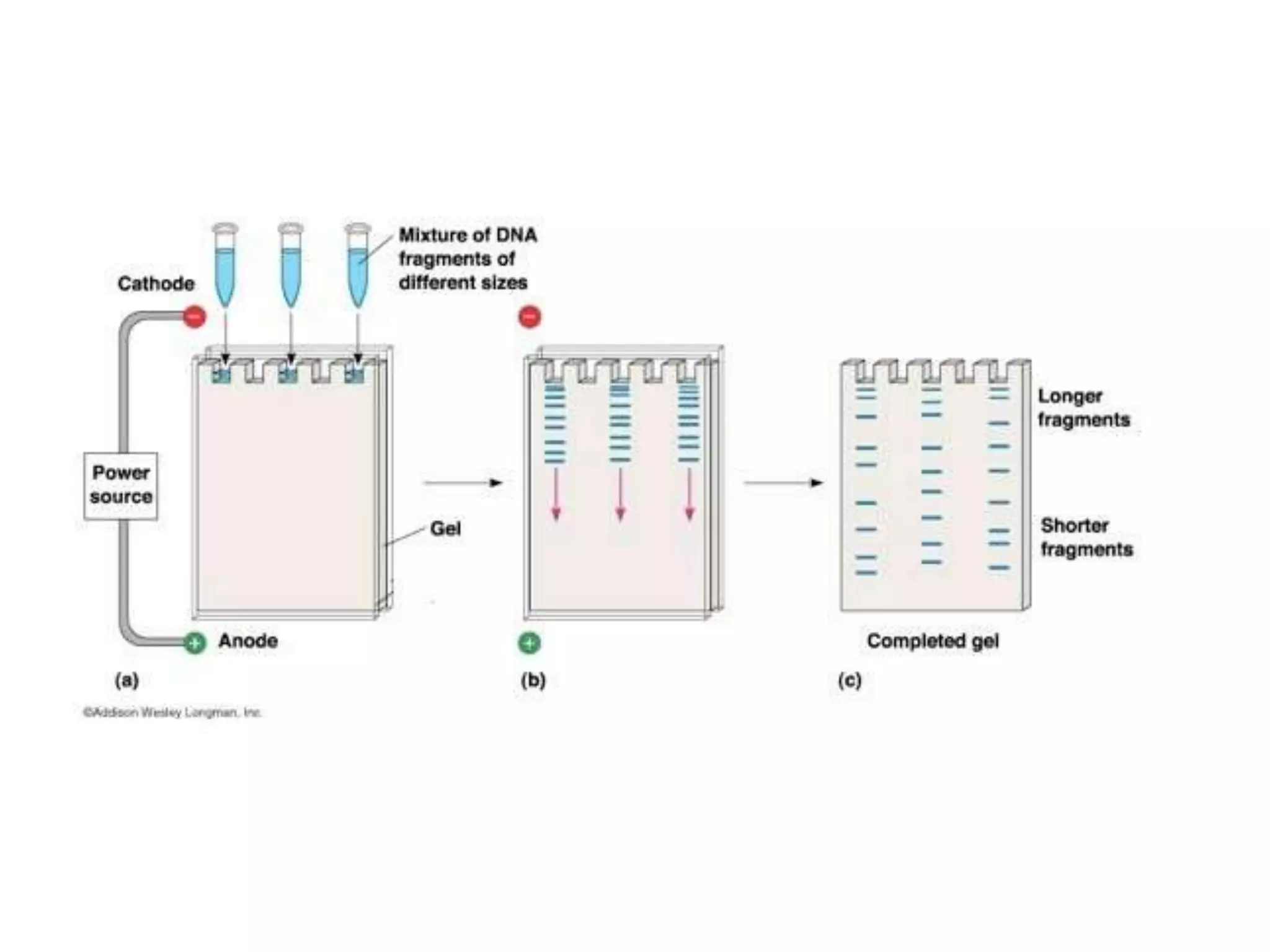
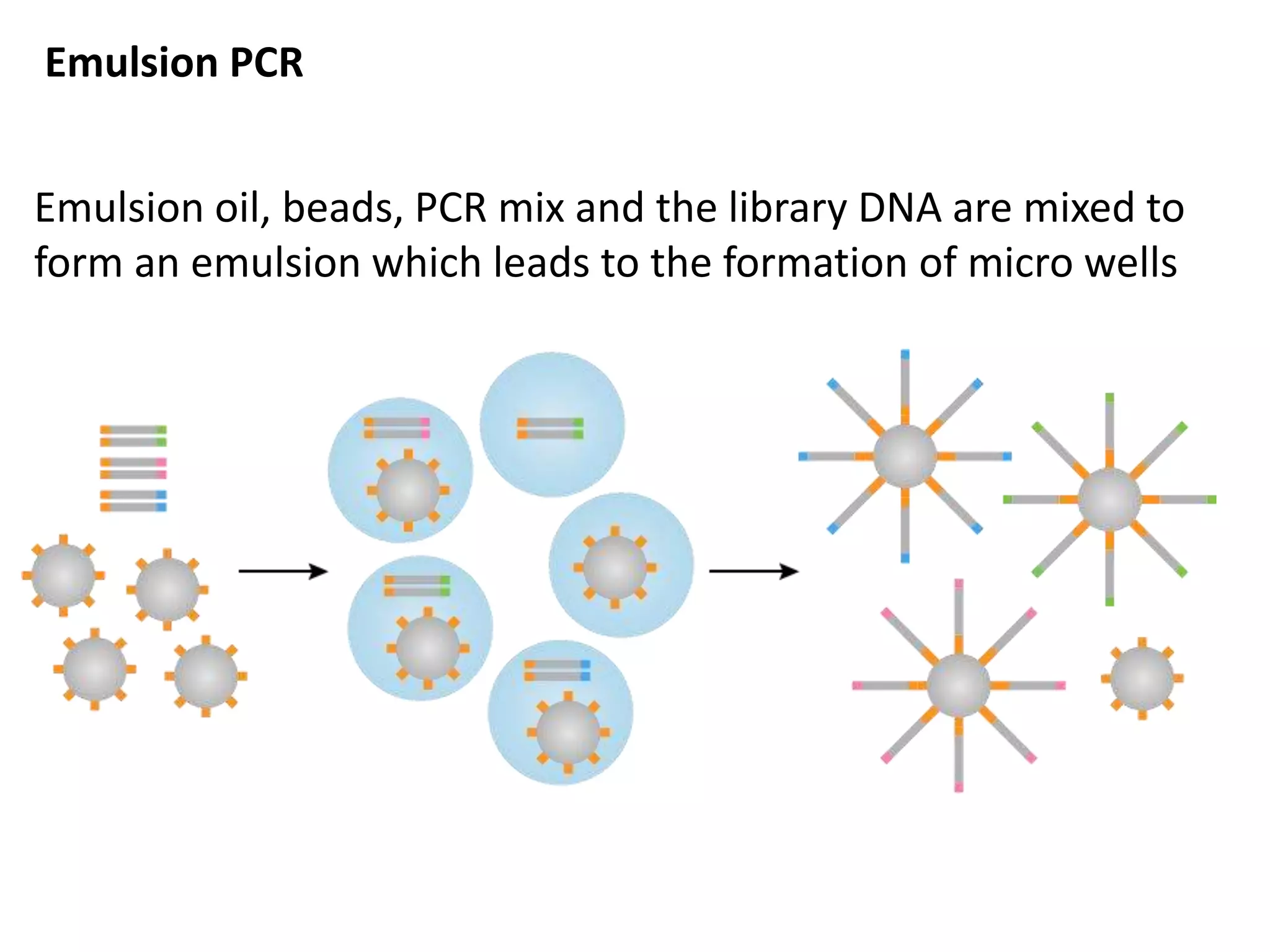
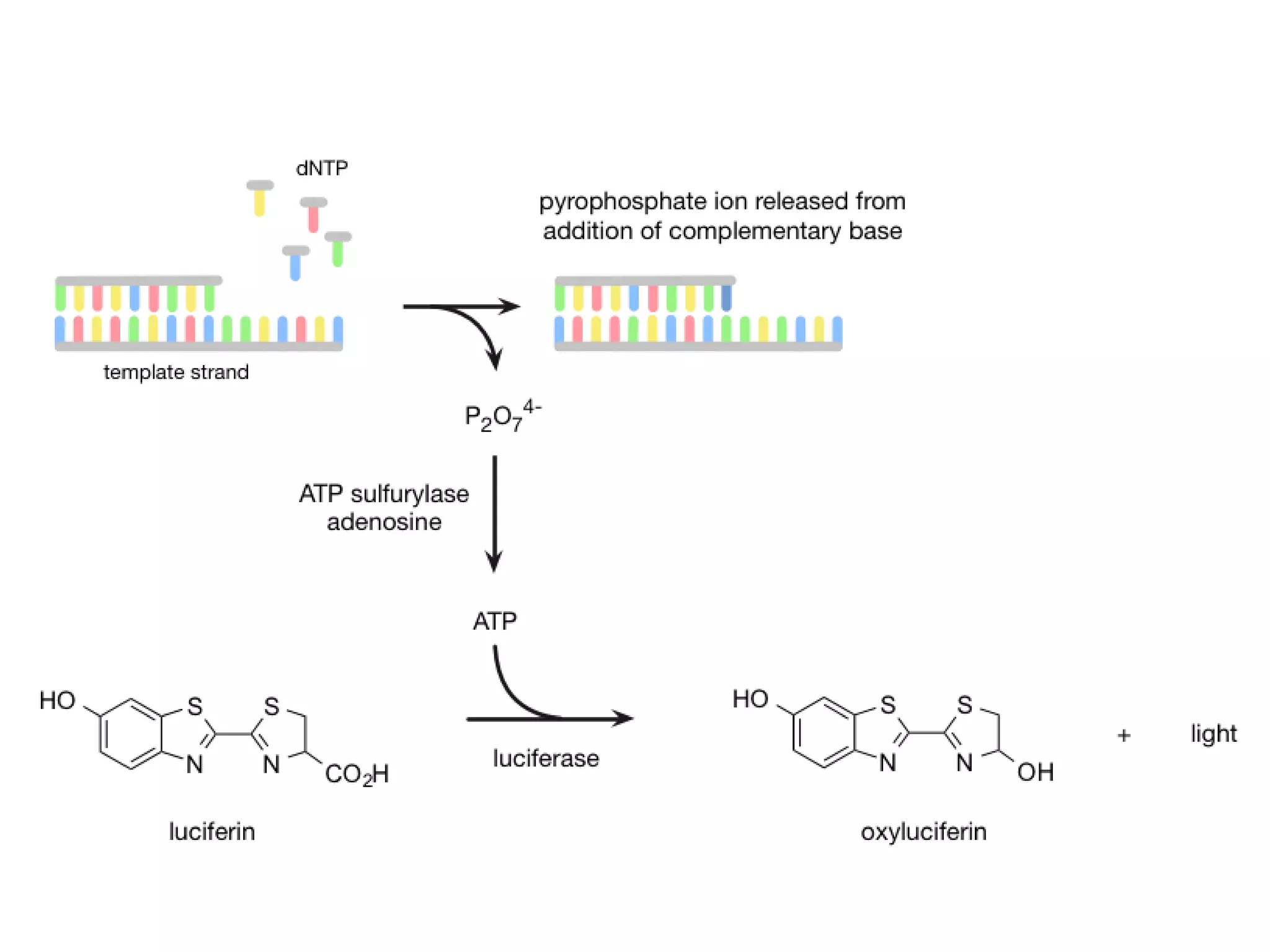
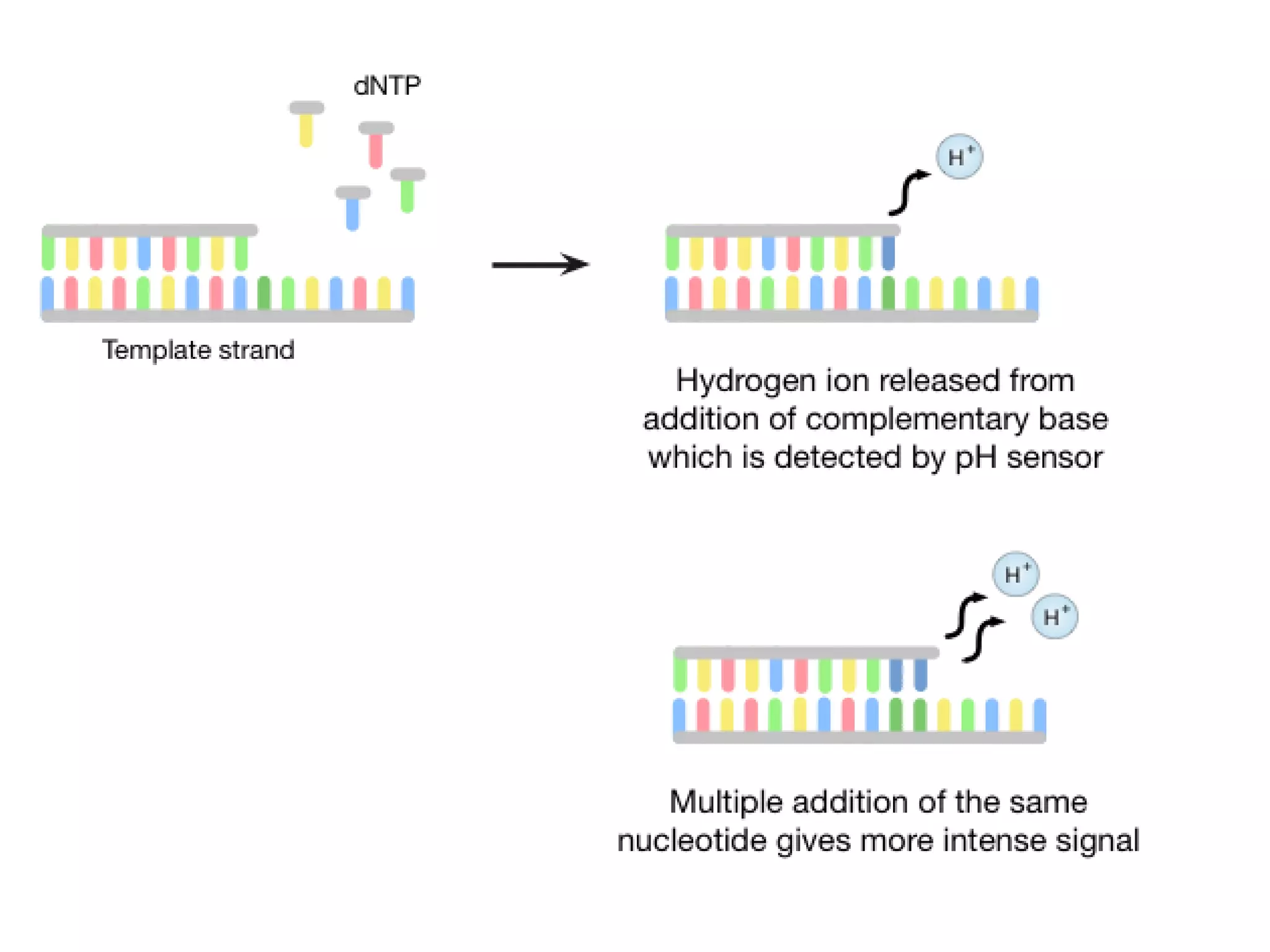
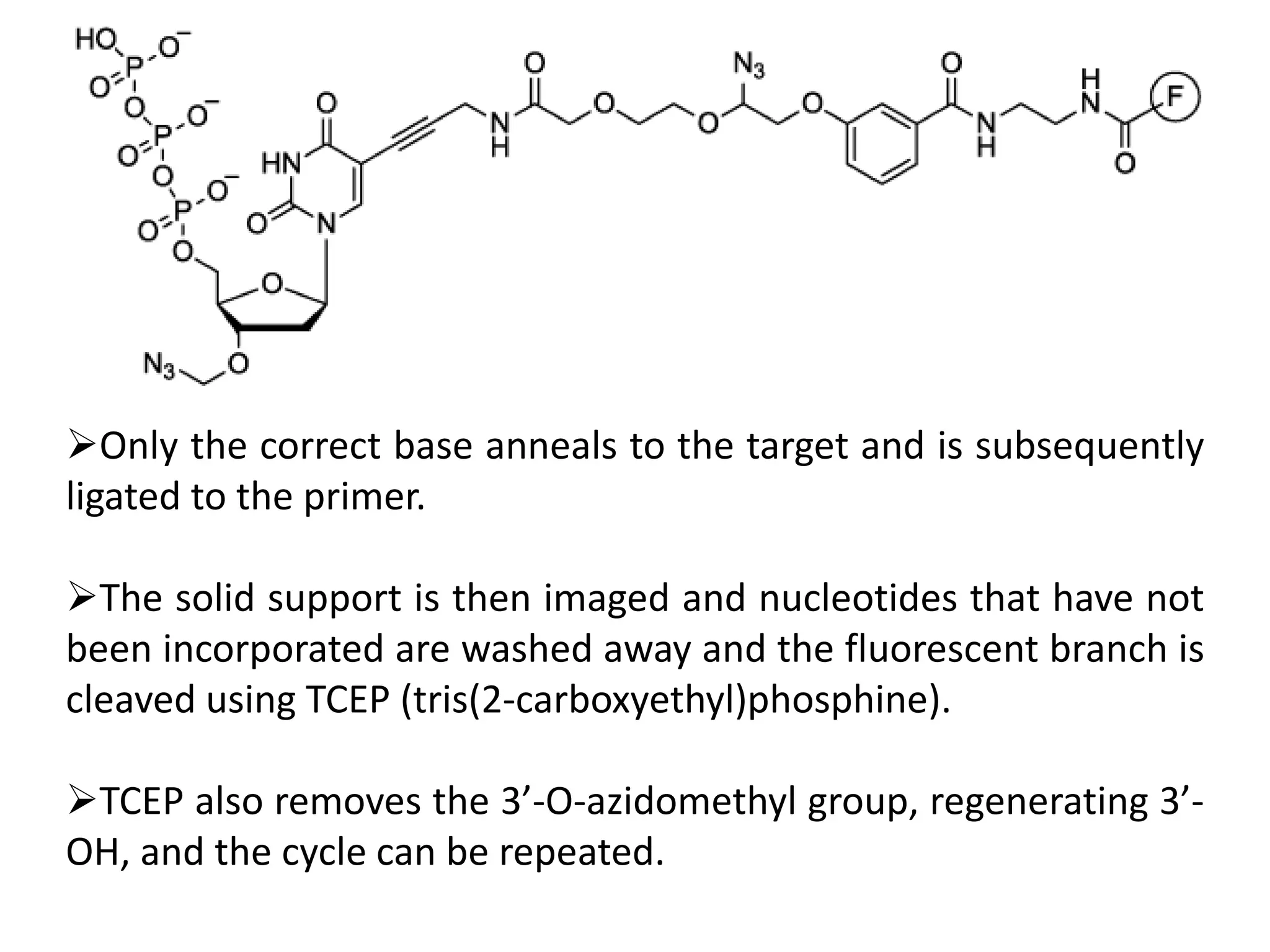
The document discusses DNA sequencing, which is the process of determining the order of nucleotides in a DNA strand, essential for genome analysis. It outlines historical advancements in sequencing techniques, including Sanger sequencing and Maxam-Gilbert sequencing, as well as introducing Next Generation Sequencing (NGS) methods like 454 pyrosequencing, Ion Torrent, and Illumina. Each method varies in its approach to sequencing, providing insights into the evolving capabilities and technologies used in genetic research.