Downloaded 14 times









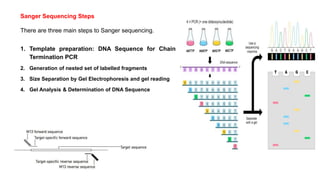

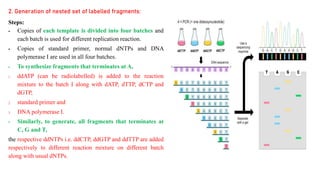




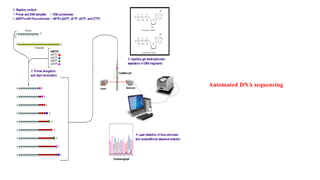



DNA sequencing refers to determining the order of nucleotides in a DNA molecule. The first DNA sequence was obtained in the 1970s using chromatography. Modern methods use dye-based sequencing and automation. The two main historical methods are the Maxam-Gilbert chemical degradation method and the Sanger dideoxy chain termination method. Next generation sequencing now allows millions of DNA molecules to be sequenced in parallel through massively parallel sequencing technologies.

















































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)





