
Downloaded 46 times

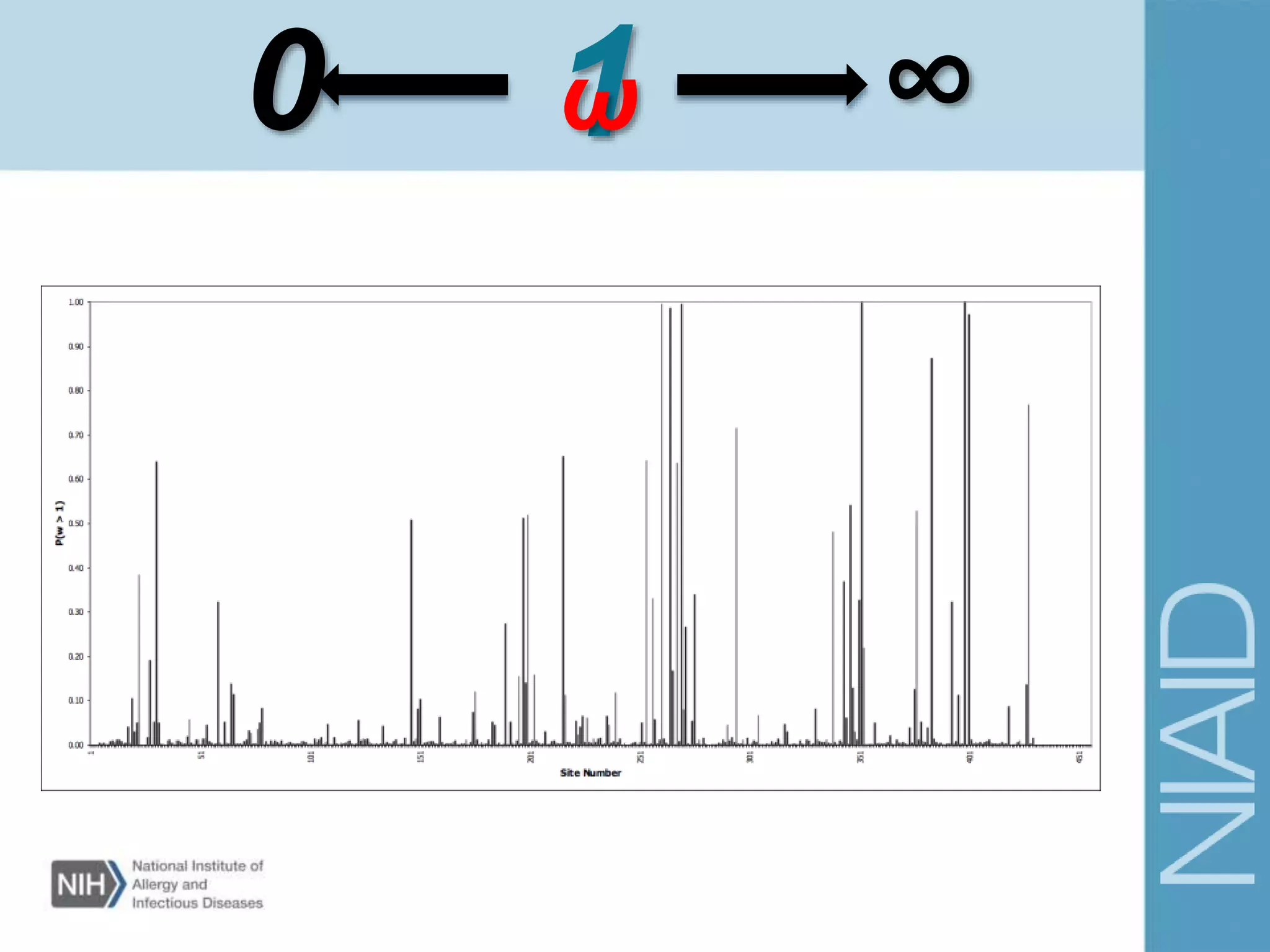




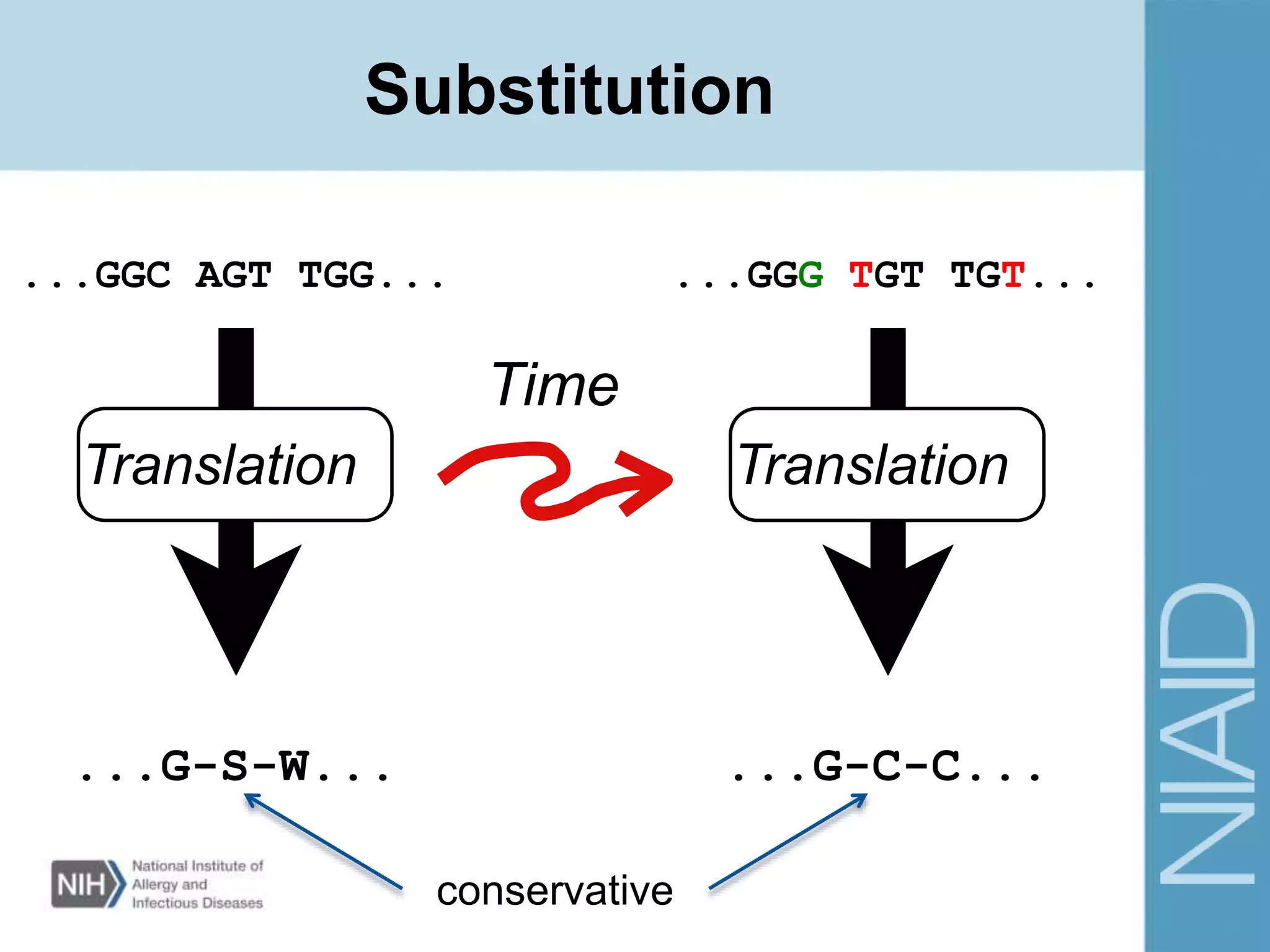








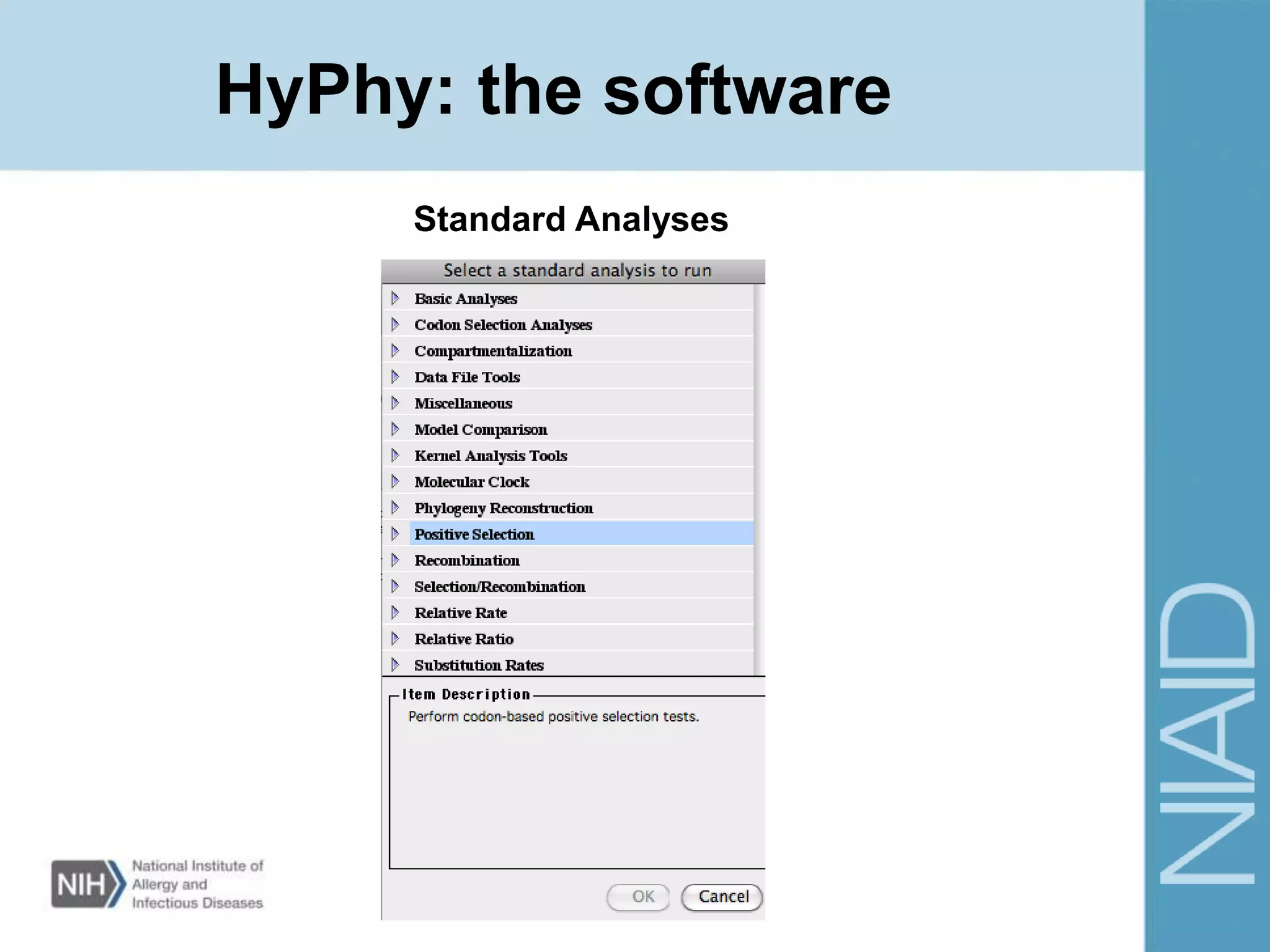
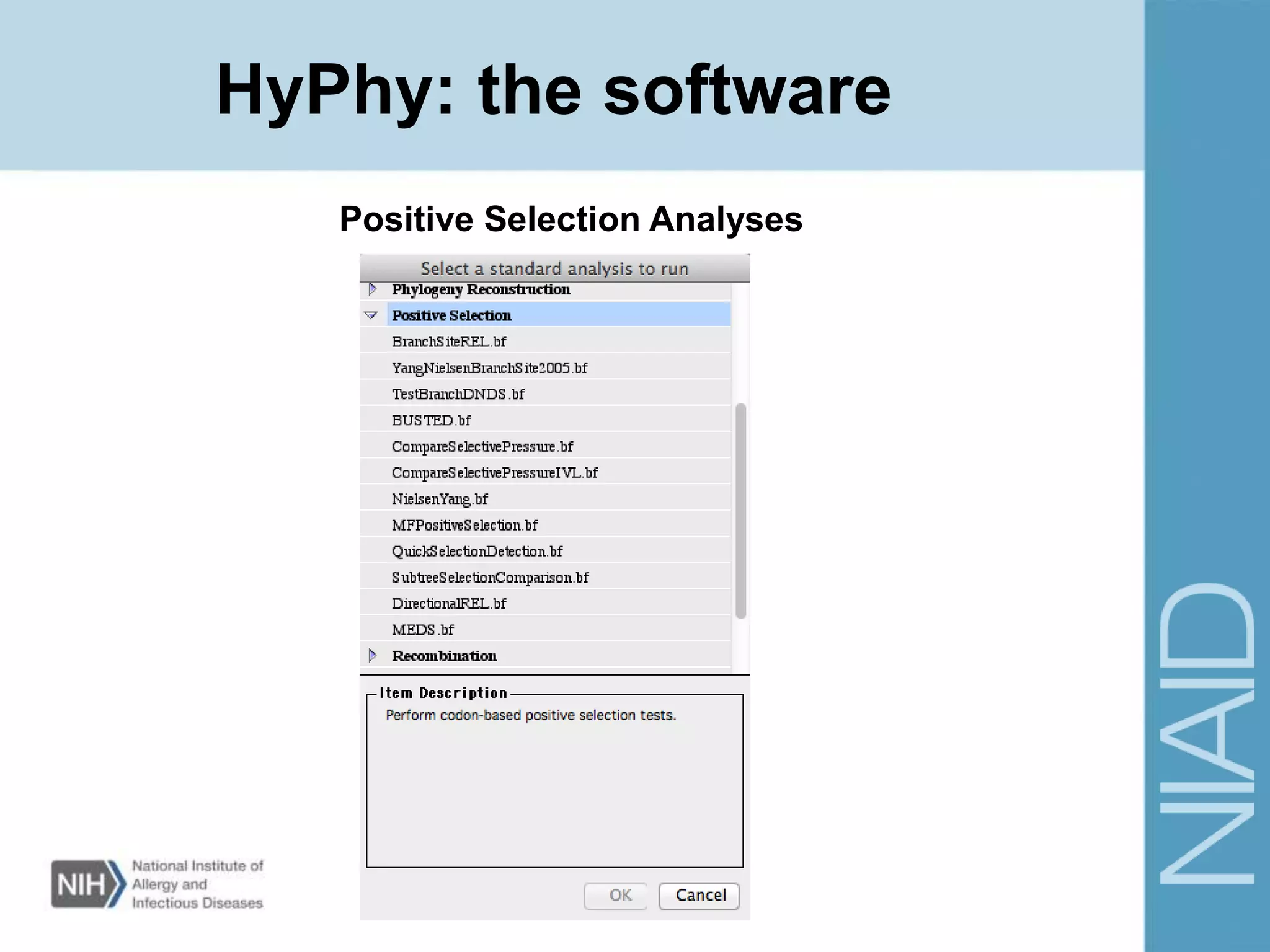


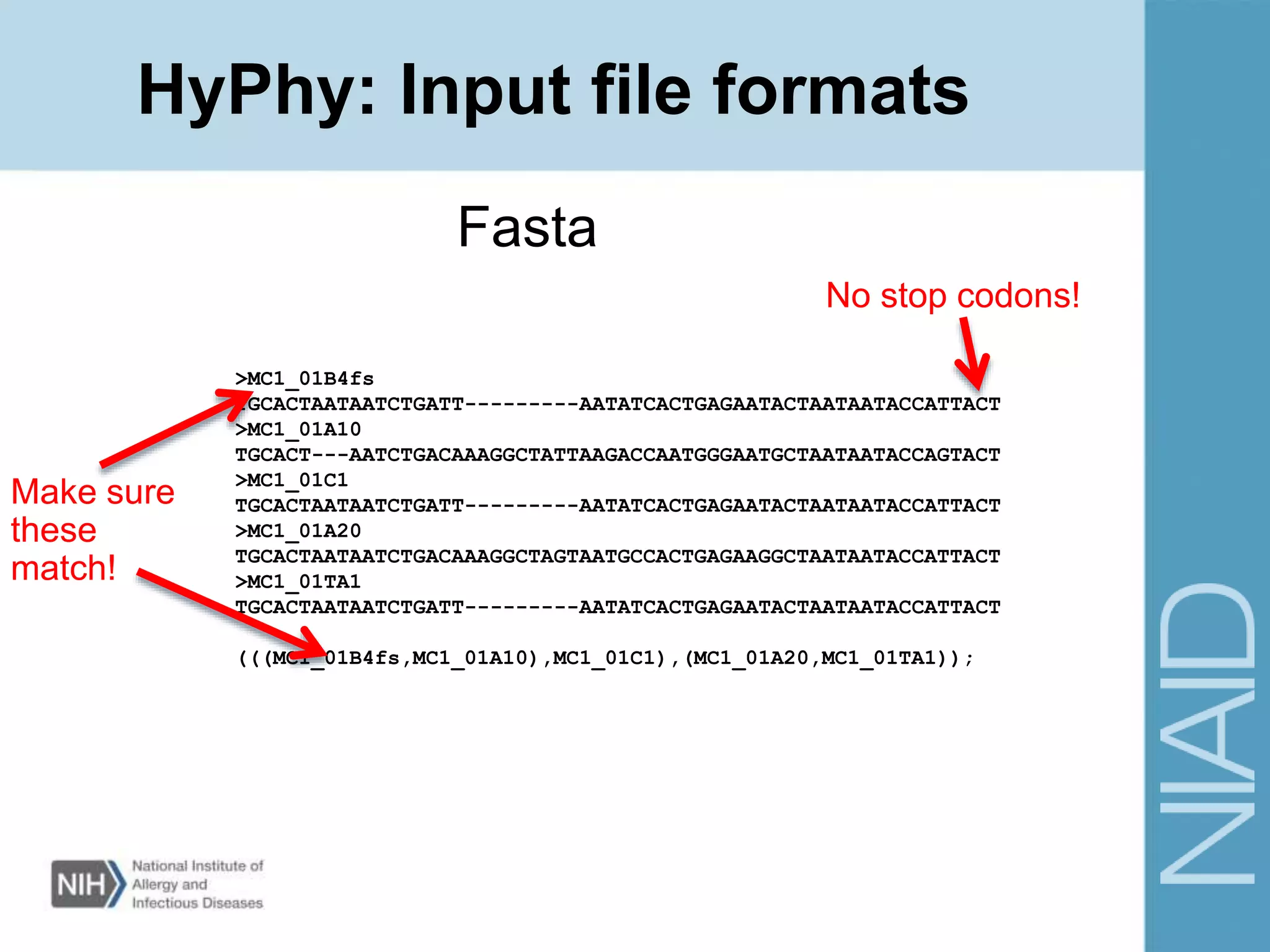








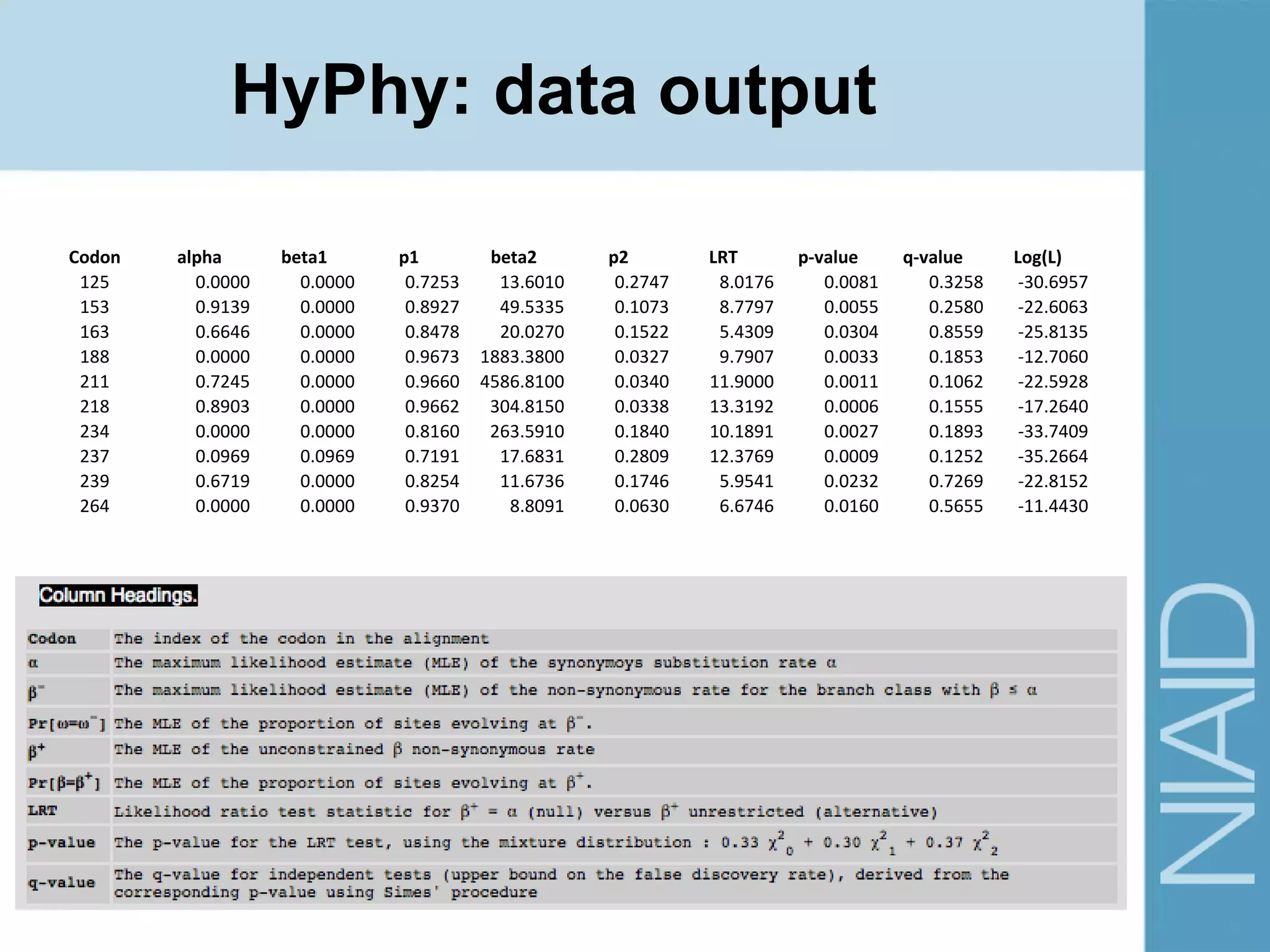
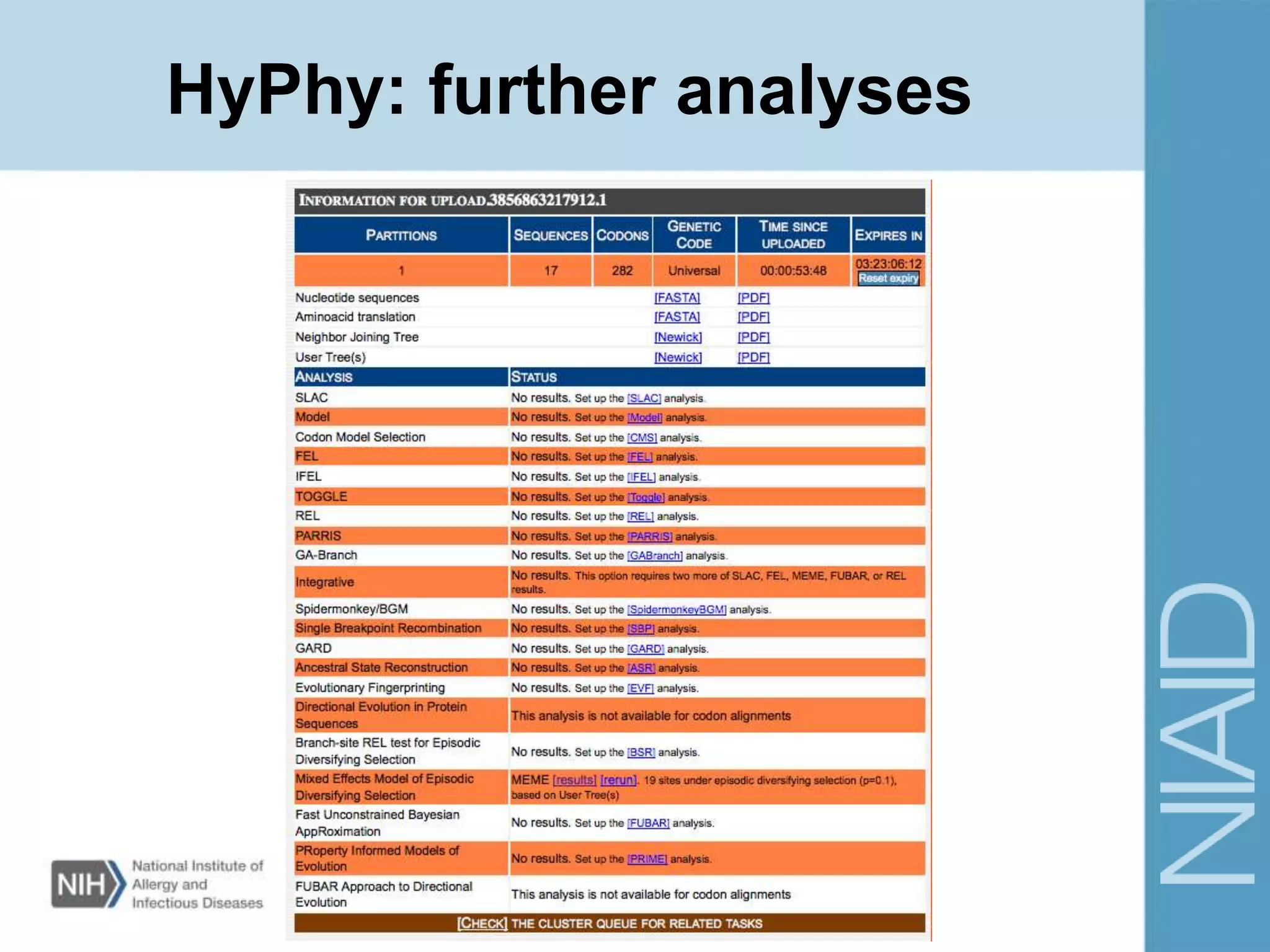








This document provides an overview of selection analysis using the HyPhy software. It discusses different types of selection including positive, purifying, and neutral selection. The document explains how HyPhy can be used to calculate dN/dS ratios and quantify selection at individual sites or along lineages. It provides information on HyPhy input formats and how to prepare sequence data and phylogenetic trees. The document also outlines standard selection analyses in HyPhy including the REL, FEL and MEME models and how to interpret the output.







































































































