The document discusses the maximum parsimony method for constructing phylogenetic trees. It states that this method minimizes the number of evolutionary changes needed to explain the differences between sequences. The method prefers the simplest phylogenetic tree that requires the fewest evolutionary changes between ancestral and descendent sequences. It also discusses evaluating different possible trees based on the total number of changes needed across all sequence positions to identify the most parsimonious tree.
Introduction of the topic of Maximum Parsimony presented by Shruthi K, a student of Microbiology.
Introduction to phylogenetic trees, evolutionary events, and parsimony methods that minimize variations in common ancestral sequences.
Difference between rooted and unrooted trees, discussion of external nodes, internal nodes, and tree topology.
List of online resources and software packages relevant for phylogenetic analysis in microbiology.
Explanation of character-based methods over distance methods, focusing on Maximum Parsimony.
Discussion of how parsimony selects trees with the fewest evolutionary changes based on Occam’s razor concept.
Details on multiple sequence alignment, the categorization of informative and uninformative sites, and analysis of unrooted trees.Evaluation of trees for their evolutionary changes, advantages of parsimony such as simplicity and methodological insights.
Disadvantages of the parsimony method, such as long-branch attraction and inconsistency in high mutation rates.Explanation of long branch attraction and how to mitigate its effects in phylogenetic analysis.
List of references and resources for further reading on bioinformatics and phylogenetics.
Phylogenetic trees,or evolutionary trees, are the basic structures
necessary to examine the relationships among organisms.
They model evolutionary events of vertical and horizontal descent.
The parsimony method is one such approach where it minimises the
number of steps to generate variations from common ancestral
sequences.
It prefers simplest explanation over more complex explanations.
A multiple sequence alignment (msa) is required to predict which
sequence positions are likely to correspond.
3.
For eachaligned position, phylogenetic trees that require the
smallest number of evolutionary changes to produce the observed
sequence changes from ancestral sequences are identified.
Finally, those trees that produce the smallest number of changes
overall for all sequence positions are identified.
McLennan, D.A. Evo Edu
Outreach (2010) 3: 506.
https://doi.org/10.1007/s12052-
010-0273-6
4.
A rootedtree is used to make inferences about the most common
ancestor of the leaves or branches of the tree. Most commonly the
root is referred to as ‘outgroup’.
An unrooted tree is used to make an illustration about the leaves or
branches, but not make assumption regarding a common ancestor.
V.K., Singh & Singh, Anil &
Kayastha, Arvind & Singh,
Brahma. (2014). Legumes in
the Omic Era. 10.1007/978-1-
4614-8370-0_12.
5.
External nodes:things under comparison; operational
taxonomic units (OTUs).
Internal nodes: ancestral units; hypothetical; goal is to
group current day units.
Topology: branching pattern of a tree.
Branch length: amount of difference that occurred along
a branch.
Monophyletic group, or clade, is a group of organisms
that consists of all the descendants of a common
ancestor.
6.
Entrez: www.ncbi.nlm.nih.gov/Taxonomy
Ribosomal database project: rdp.cme.msu.edu/html/
Tree of Life:
phylogeny.arizona.edu/tree/phylogeny.html
PHYLLIP PACKAGE:
i. DNAPERS
ii. DNAPENNY – For more sequences
1. DNACOMP – finds tree that supports largest number
of sites.
2. DNAMOVE – interactive analysis of parsimony
7.
Tree oflife: Analyzing changes that have occurred in
evolution of different organisms.
Phylogenetic relationships among genes can help
predict which ones might have similar functions (e.g.,
ortholog detection).
Follow changes occuring in rapidly changing species
(e.g., HIV virus)
8.
This isan example of character based method.
They are based on sequence character rather than
pairwise distances.
They count mutational events accumulated on the
sequences and may therefore avoid loss of information
when character is converted to distances.
Thereby evolutionary dynamics can be studied and
ancestral approaches can also be studied.
Maximum parsimony is an example for this method.
9.
The parsimonymethod chooses a tree that has fewest
evolutionary changes or mutations or shortest overall
branch length.
Based on Occam’s razor philosophy.
Reduces chances of inconsistencies, ambiguities and
redundancies.
By minimizing the changes, the method minimizes
the phylogenetic noise owing to homoplasy and
independent evolution.
10.
•The four-way multiple
sequencealignment contains
positions that fall into two
categories – informative and
uninformative sites.
• For the first position all four
sequences have same character
and no mutations- invariant
• Position 2 and 4 have
minimum two mutations
which are derived from
ancestors - informative
12.
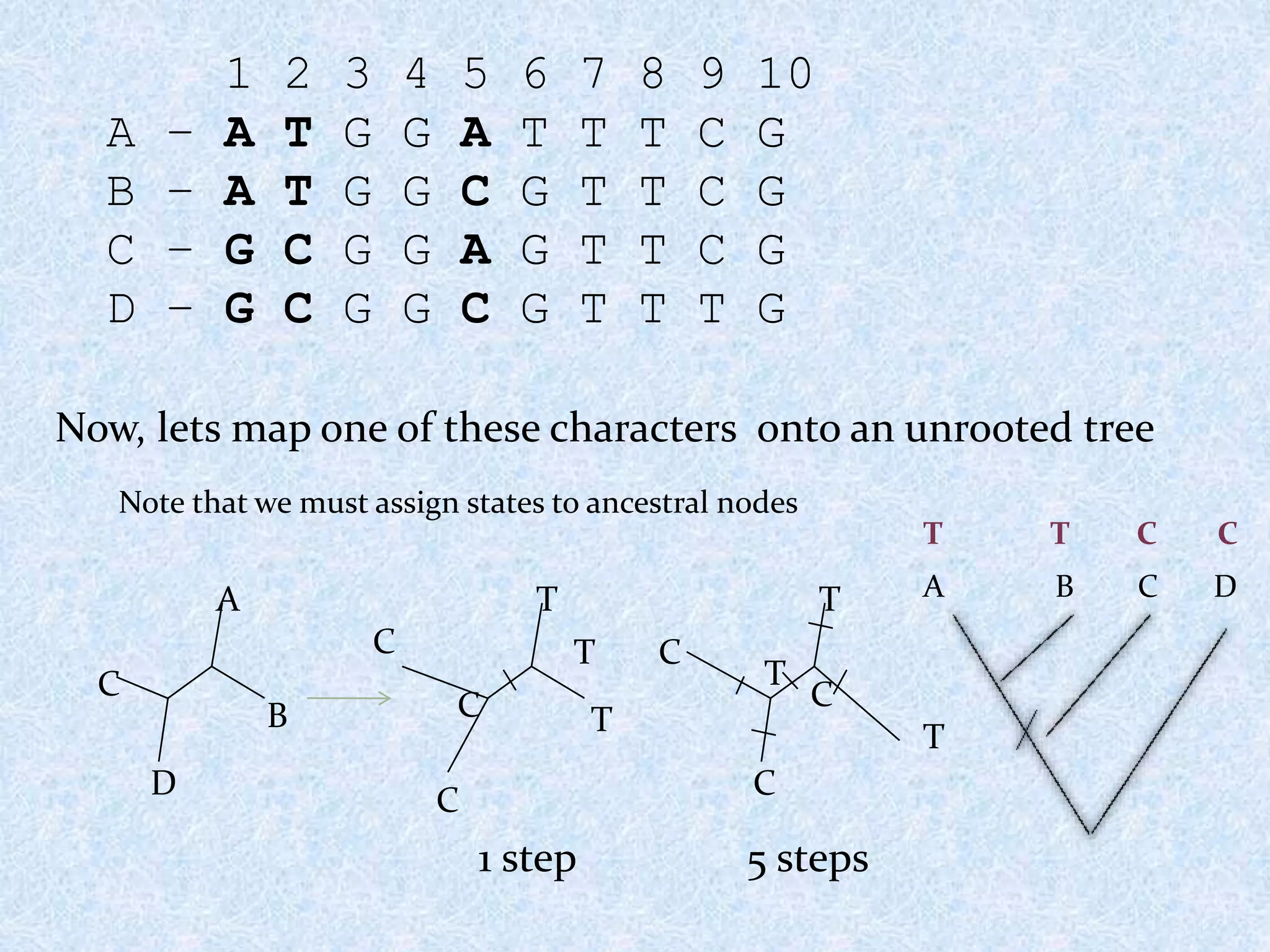
1 2 34 5 6 7 8 9 10
A – A T G G A T T T C G
B – A T G G C G T T C G
C – G C G G A G T T C G
D – G C G G C G T T T G
Now, lets map one of these characters onto an unrooted tree
Note that we must assign states to ancestral nodes
A
D
B
C
T
C
T
C T
C
1 step
T
C
T
C
C
T
5 steps
A B C D
T T C C
13.
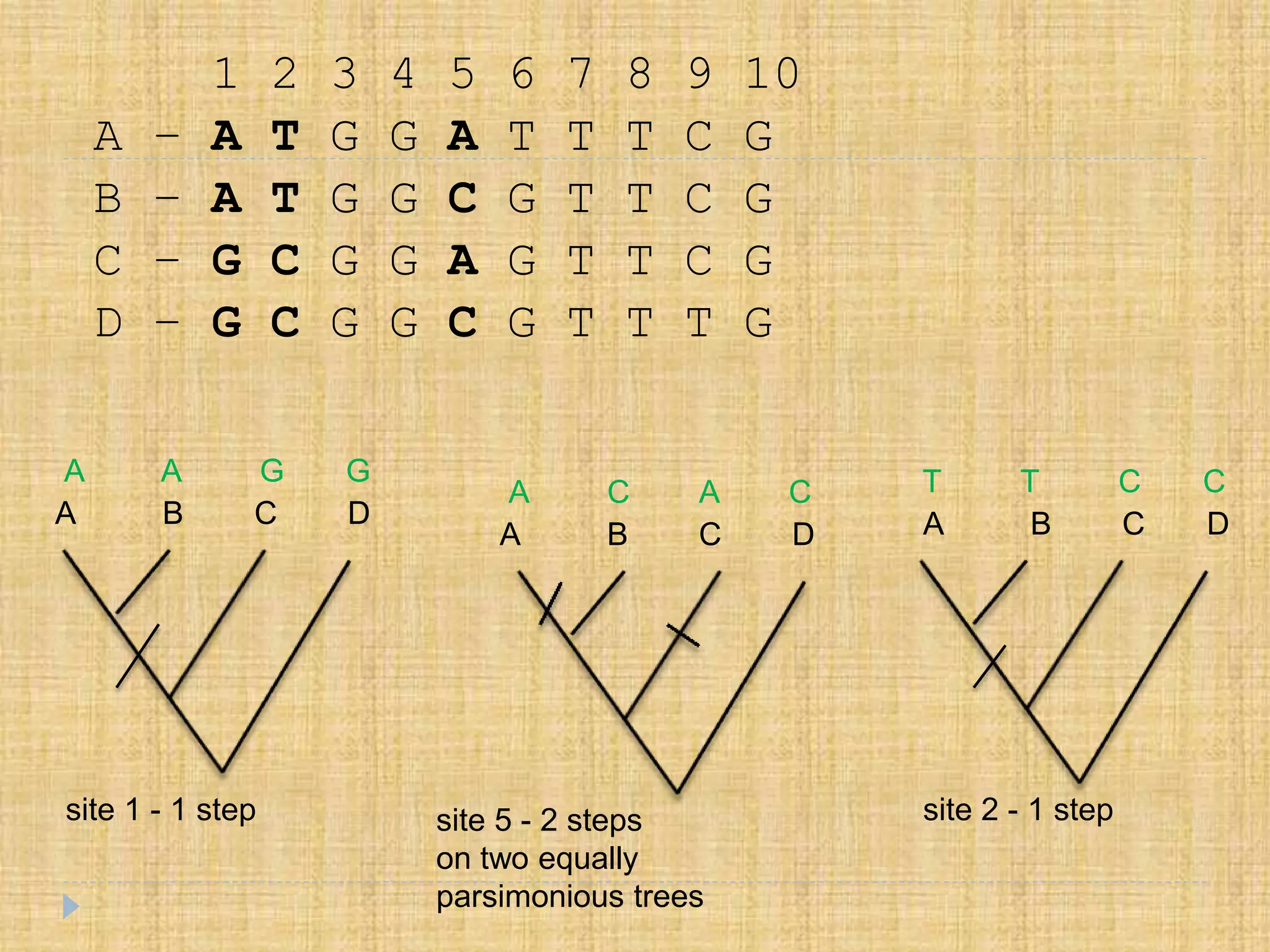
1 2 34 5 6 7 8 9 10
A – A T G G A T T T C G
B – A T G G C G T T C G
C – G C G G A G T T C G
D – G C G G C G T T T G
site 1 - 1 step
A B C D
A B C D A B C D
A A G G
A C A C T T C C
site 5 - 2 steps
on two equally
parsimonious trees
site 2 - 1 step
14.
Mapping should alsobe done for all other sites
Sites 3,4,7,8,10 – 0 steps
Mapping should also be done for all possible trees
site 6 – 1 step
1 2 3 4 5 6 7 8 9 10
A – A T G G A T T T C G
B – A T G G C G T T C G
C – G C G G A G T T C G
D – G C G G C G T T T G
G
T
G
G
G
G
C
T
C
C
C
C
site 9 - 1 step
15.
There are threepossible unrooted trees for four taxa.
B
C
D
A
A
B
D
C
A
D
B
C
((A,B),(C,D)) ((A,D),(C,B)) ((A,C),(B,D))
16.
CTND…
Evaluate eachpossible tree for all sites to determine
the smallest total number of changes necessary to
generate each one
Note sites 3,4,6,7,8,9,10 are the same for every tree –
parsimony uninformative
Sites
Tree 1 2 3 4 5 6 7 8 9 10 Total
((A,B),(C,D)) 1 1 0 0 2 1 0 0 1 0 6
((A,D),(C,B)) 2 2 0 0 2 1 0 0 1 0 8
((A,C),(B,D)) 2 2 0 0 1 1 0 0 1 0 7
17.
WEIGHTED PARSIMONY
Supposewe weight transversions with twice the
value of transitions
Site 5 is now weighted twice as much as sites 1
and 2
Sites
Tree 1 2 3 4 5 6 7 8 9 10 Total
((A,B),(C,D)) 1 1 0 0 4 1 0 0 1 0 8
((A,D),(C,B)) 2 2 0 0 4 1 0 0 1 0 10
((A,C),(B,D)) 2 2 0 0 2 1 0 0 1 0 8
18.
ADVANTAGES
Easy tounderstand
Makes relatively few assumptions.
Well studied mathematically
Many useful software packages
More theoretical arguments:
1. Methodologically, parsimony forces us to maximize
homologous similarity. This is not necessarily true for
other methods
2. Parsimony is based on an evolutionary assumption –
evolutionary change is rare. Not true at all for most
distance methods
19.
DISADVANTAGES
Why notuse parsimony?
Not consistent, under some scenarios it is possible (even
likely) to get the wrong tree
Long-branch attraction – similar to rate heterogeneity
problem encountered with distance methods
When DNA substitution rates are high, the probability that
two lineages will convergently evolve the same nucleotide at
the same site increases. When this happens, parsimony
erroneously interprets this similarity as a synapomorphy
(i.e., evolving once in the common ancestor of the two
lineages).
20.
VERSIONS
Versions ofparsimony
Fitch parsimony – no limitations on permissible character
changes, reversible P(A->T) = P(T->A)
Wagner parsimony – allows ordered transformations (to get
from C to G, you must proceed through A), reversible
Dollo parsimony – consider restriction site characters
P(0->1) ≠ P(1->0)
Limited non-reversibility – derived states cannot be lost
and regained
Works really well for mobile element insertion data
Camin-Sokal parsimony – evolutionary changes are
irreversible
Transversion parsimony – ignores transitions or downweights
them severely
21.
Refers tophylogenetic artifact in which rapidly
evolving taxa with long branches are placed together.
It is regardless of their true positions.
Due to assumption that all lineages evolve at the same
rate and that all mutations contribute to branch
length.
A
B D
C
Long branch
22.
The edgesleading to sequences/taxa A and C are long
relative to other branches in the tree, reflecting the
relatively greater number of substitutions that have
occurred along those two edges.
The long branch attraction occurs when rates of
evolution show considerable variation among
sequences, or where the sequences being analysed are
quite divergent.
How to overcome Long Branch Attraction?
To reduce the effects of long edges is to add
sequences/taxa that join onto those edges thus breaking
them up.
23.
Krane, Raymer.ML,Fundamental concepts of
bioinformatics, 2003, Pearson education
Xiong.J, Essential bioinformatics, 2006, Cambridge
University press.
Bioinformatics: Sequence and Genome Analysis by
Mount D., 2004 Cold Spring Harbor Laboratory Press,
New York.