Downloaded 357 times






















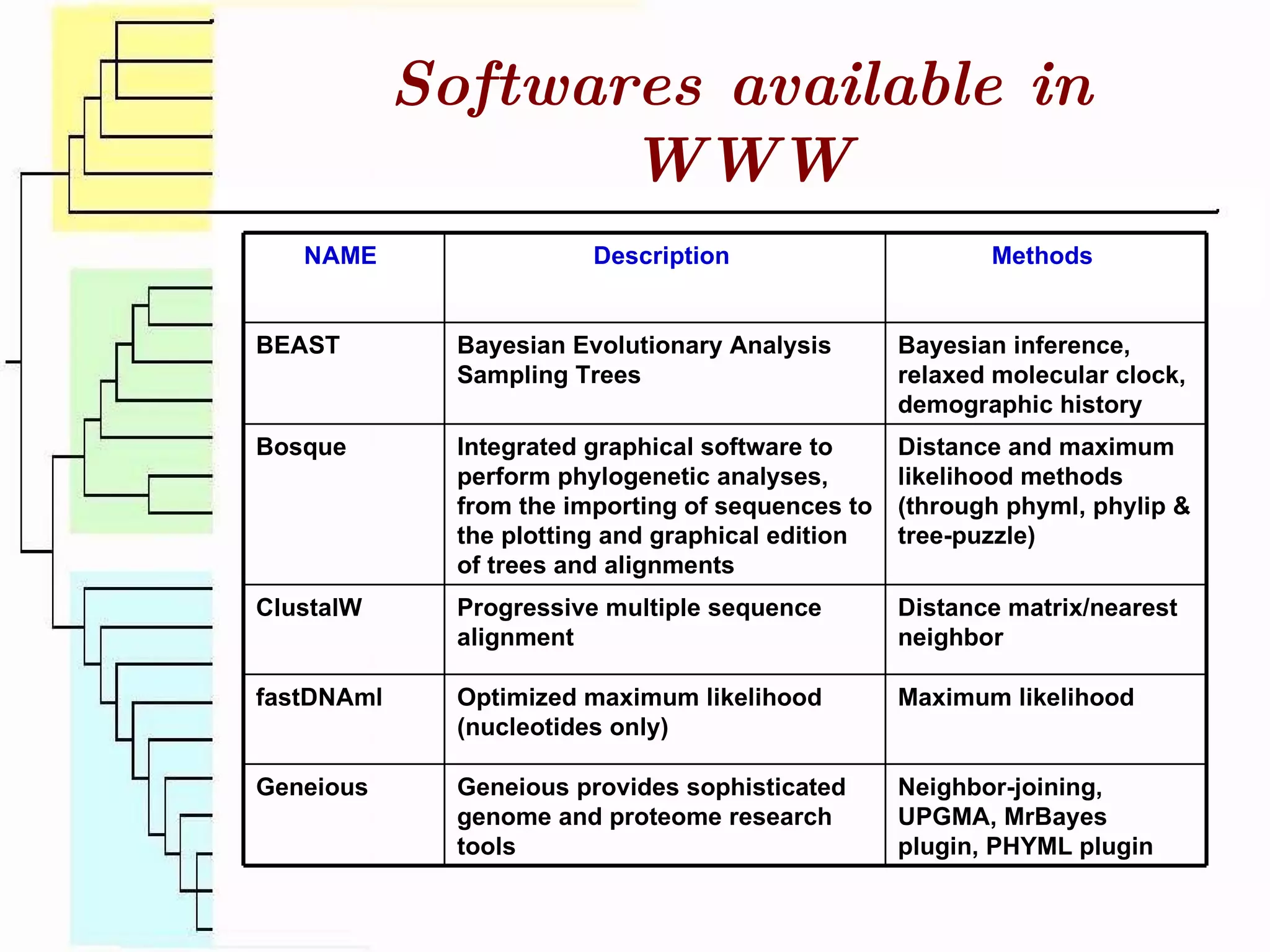
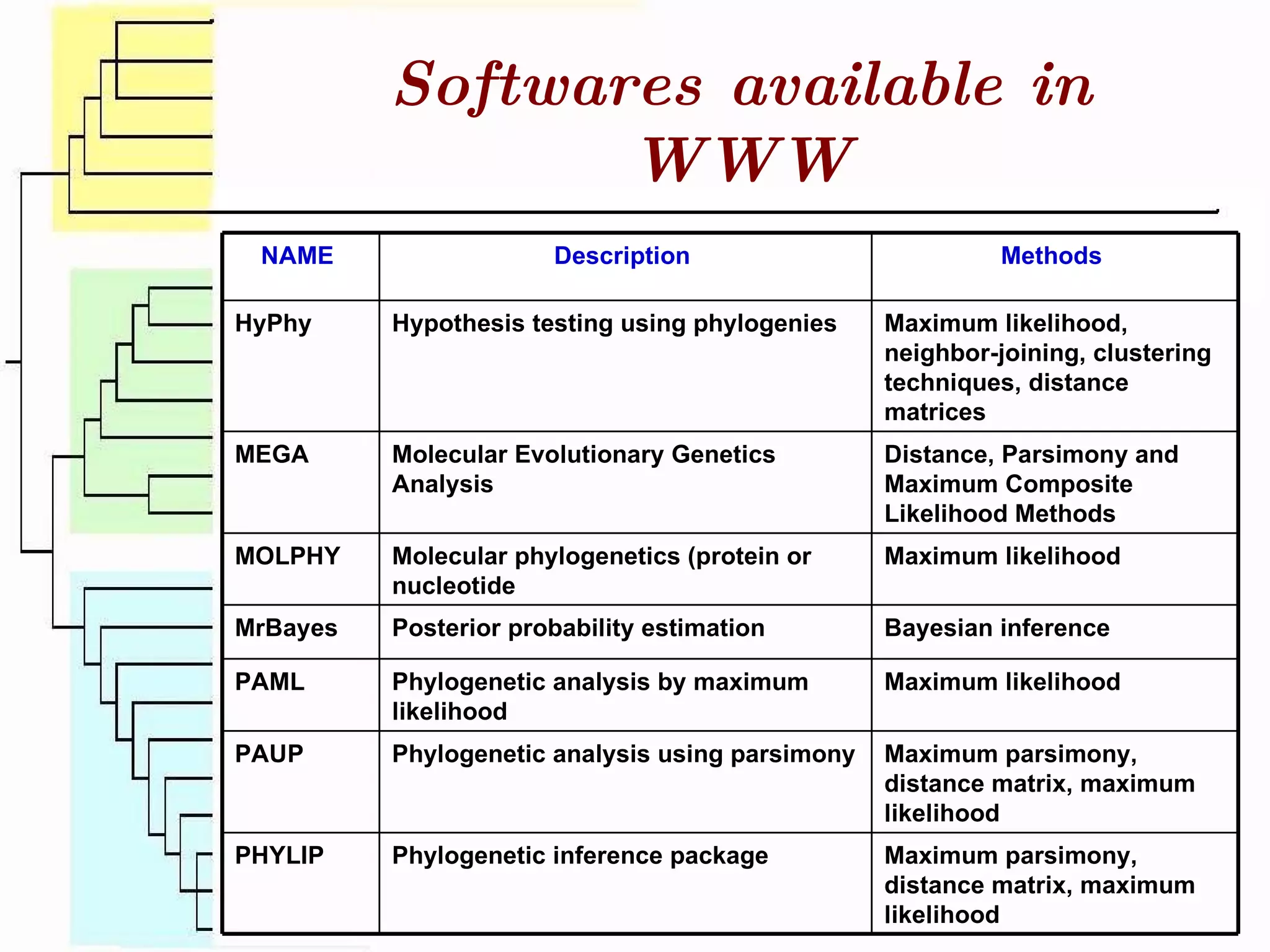
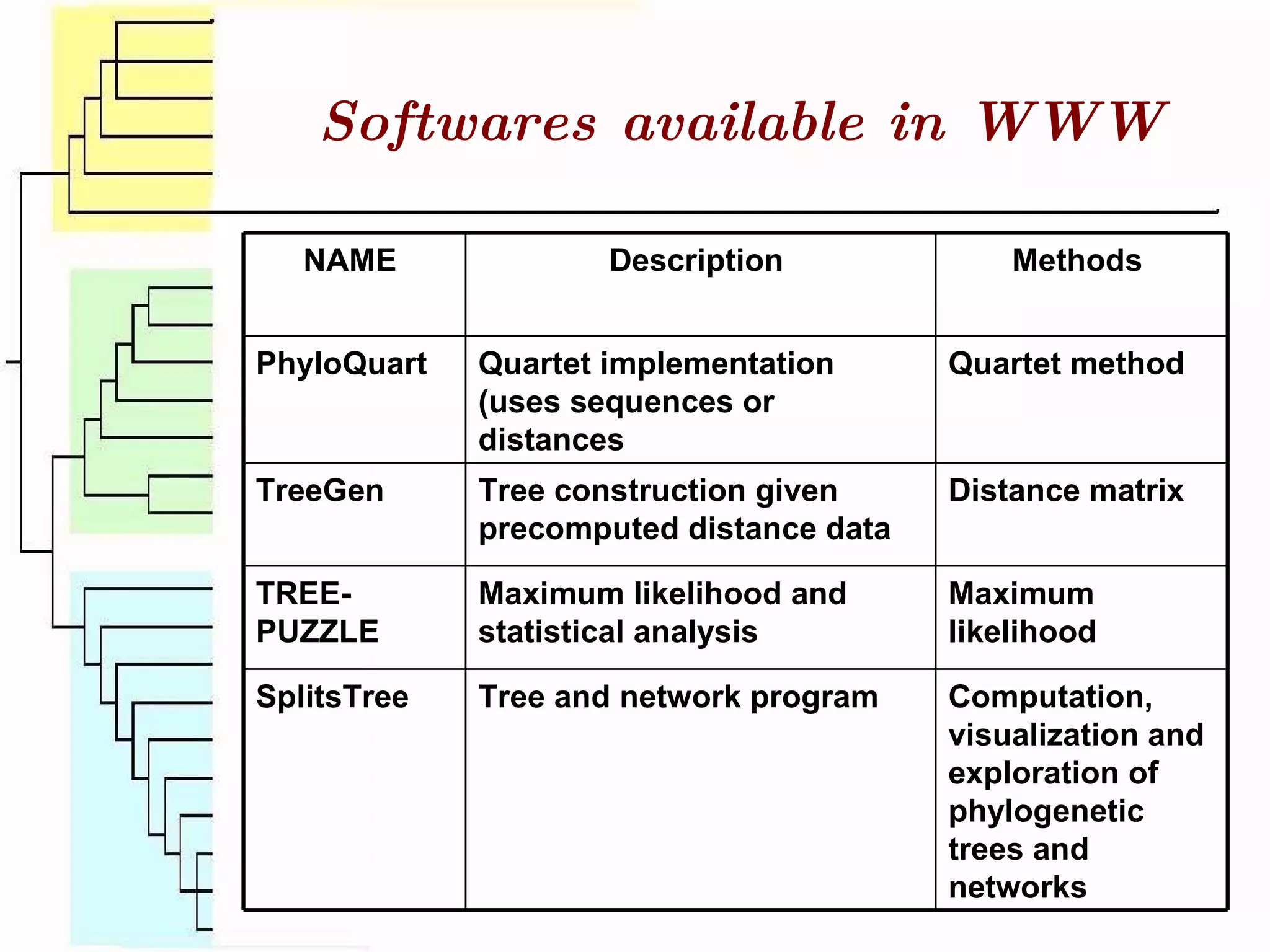

This document surveys various software programs available for phylogenetic analysis, discussing their methods, strengths, and desirable attributes. It emphasizes the importance of initial sequence alignment and selecting appropriate substitution models for accurate phylogenetic tree construction. Additionally, it outlines user requirements for the software and lists available programs along with their respective methods.










































































![Vibe Coding vs. Spec-Driven Development [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/vibecodingvsspecdrivendevelopment-251209105622-43f455e7-thumbnail.jpg?width=640&height=640&fit=bounds)











