Downloaded 1,810 times




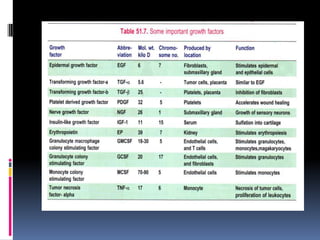

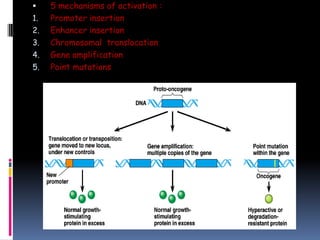



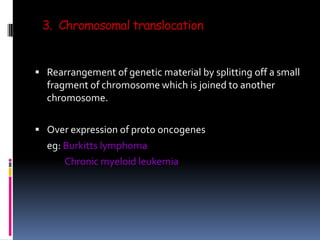
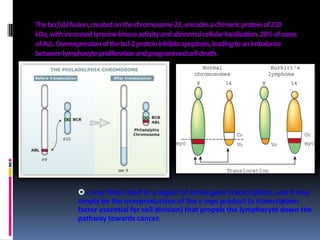
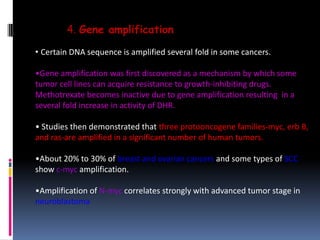

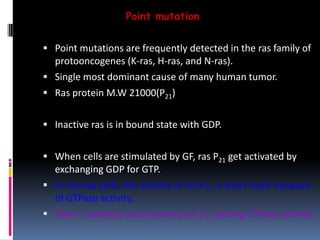
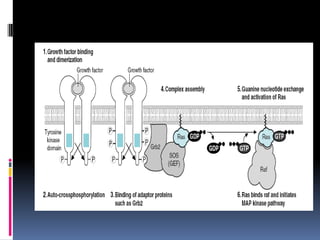
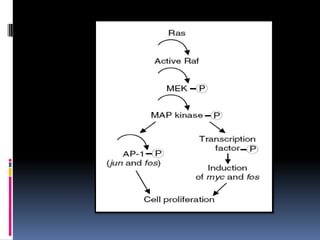


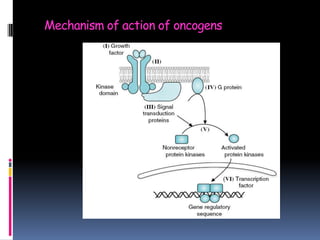
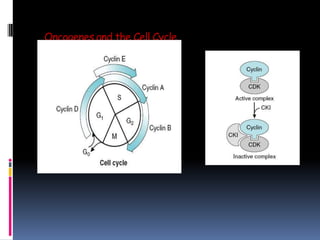



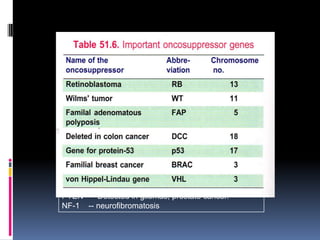

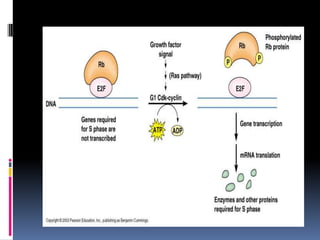

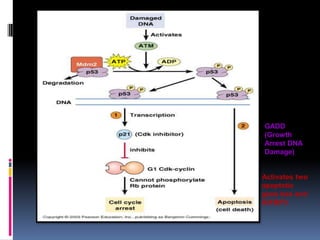

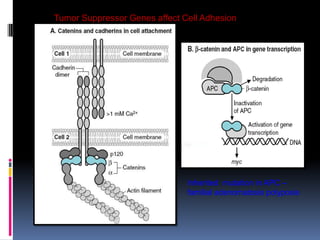


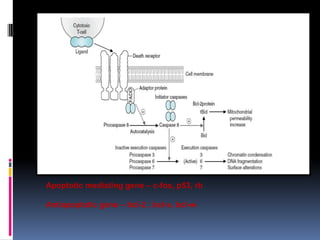
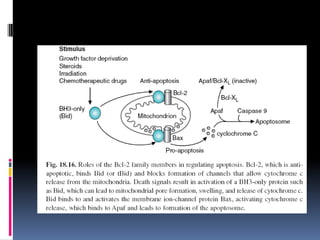




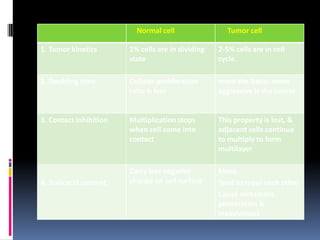
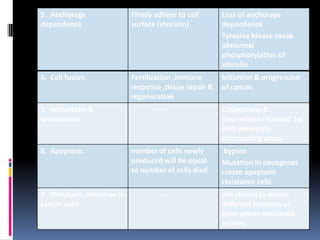


Cancer is caused by mutations in genes that regulate cell growth and proliferation. These mutations can activate proto-oncogenes into oncogenes or inactivate tumor suppressor genes. Oncogenes promote cell growth while tumor suppressor genes normally inhibit cell proliferation. Common mechanisms of proto-oncogene activation include chromosomal translocations, gene amplifications, and point mutations. Disruptions to cell cycle checkpoints, apoptosis, telomere maintenance and DNA repair pathways can also contribute to cancer development by allowing abnormal cell growth and survival.



































































































