Downloaded 311 times









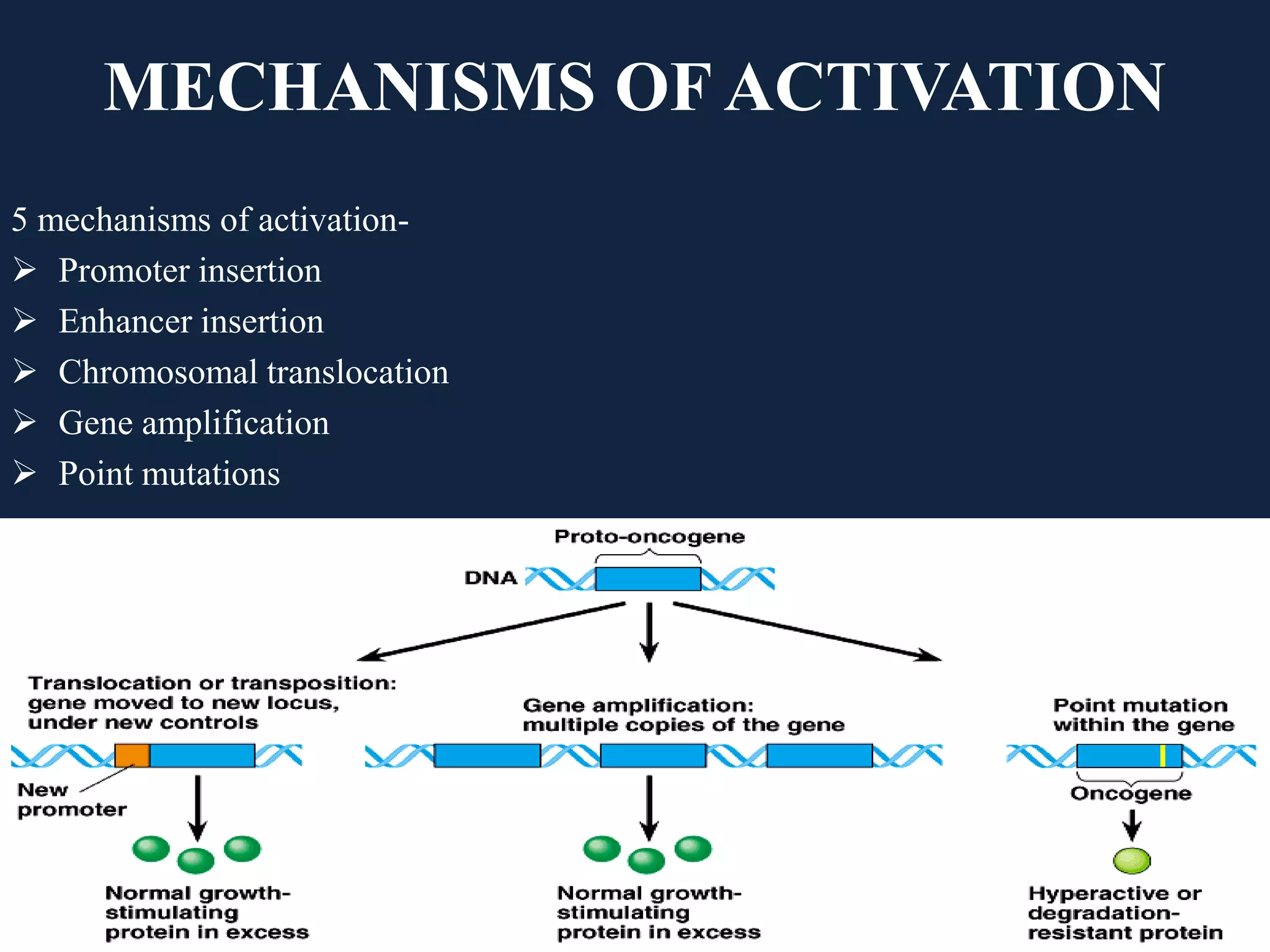
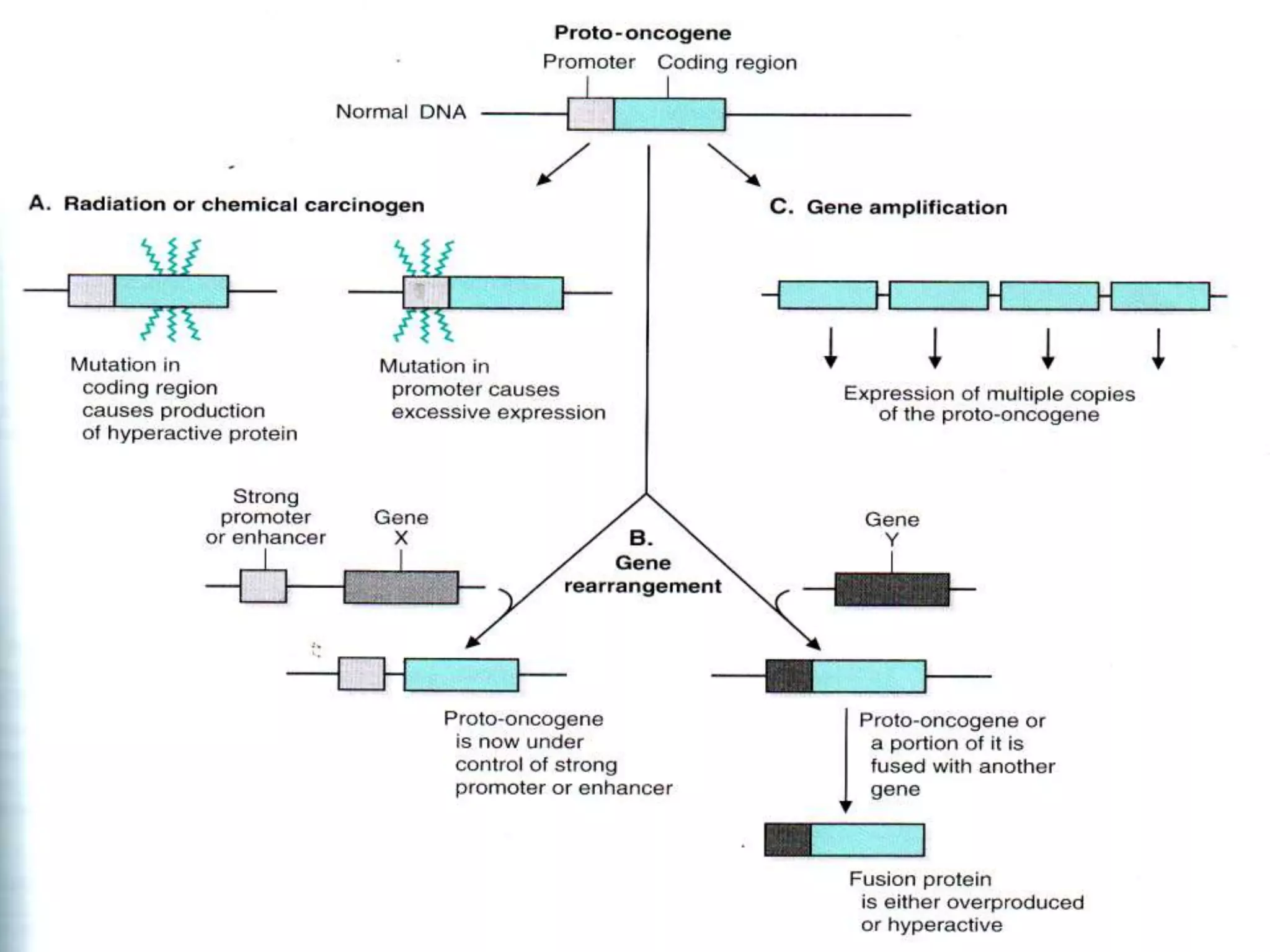



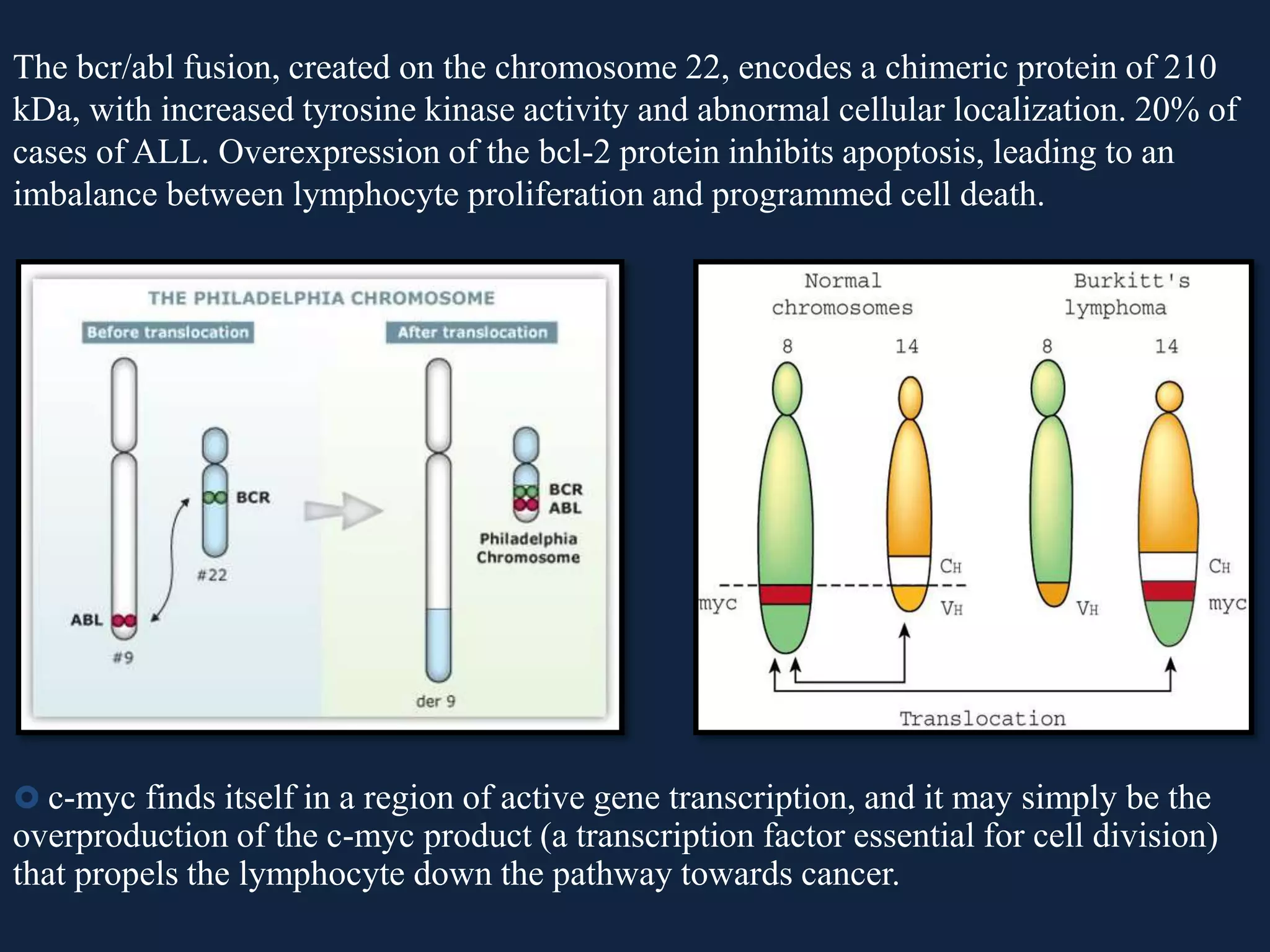




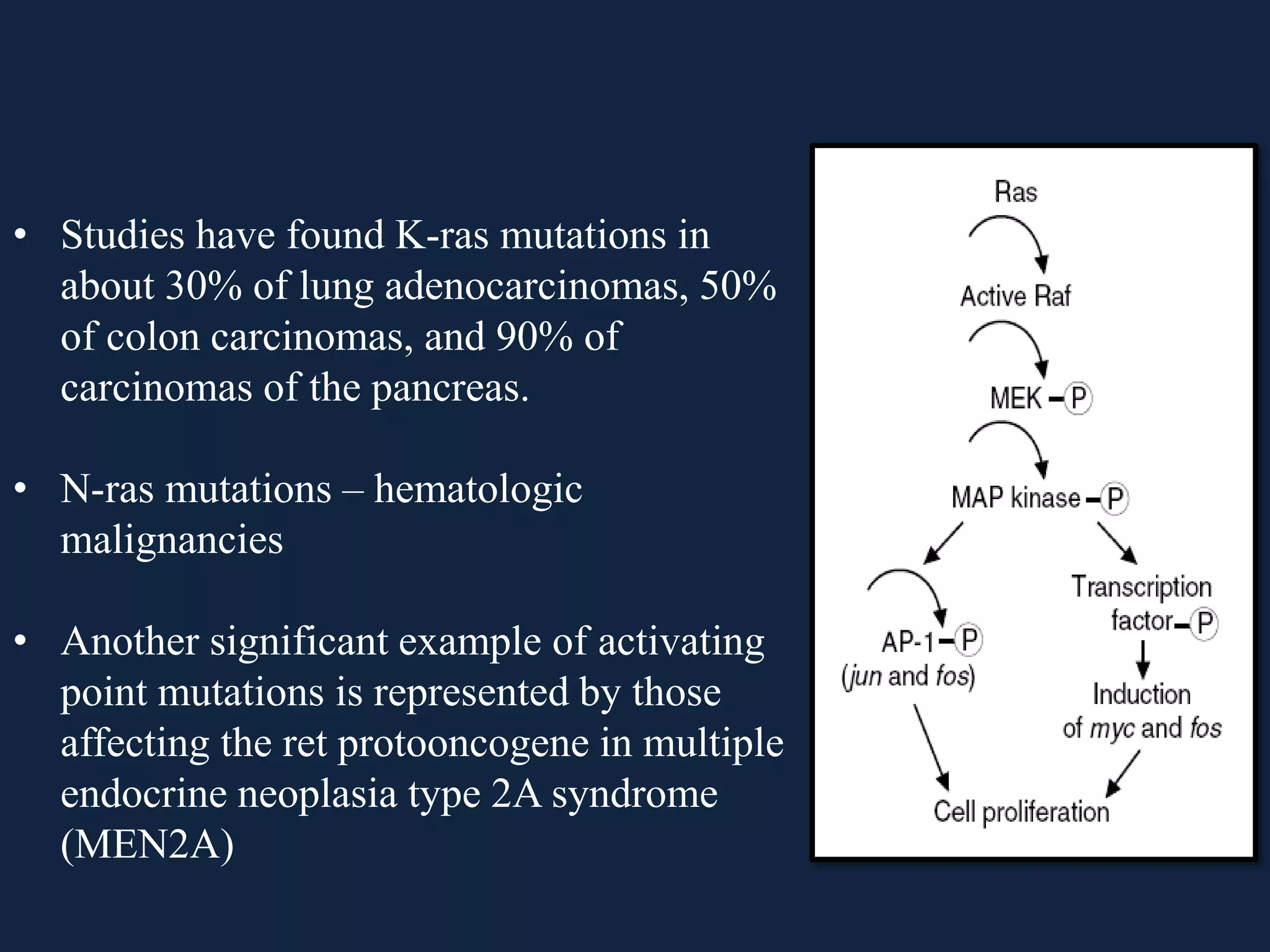


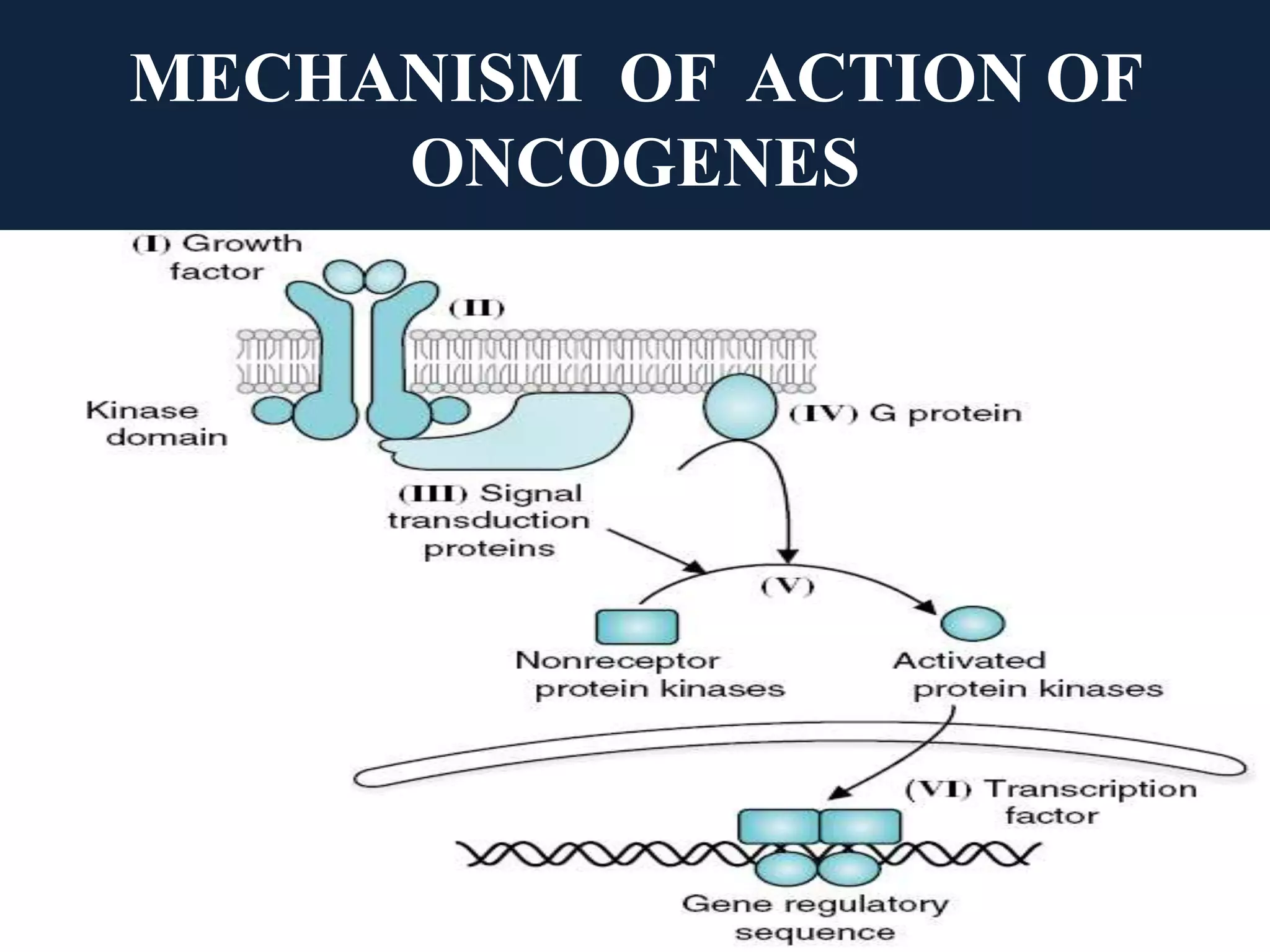









The seminar covers the role of oncogenes and protooncogenes in cancer development, emphasizing their activation mechanisms and differing roles compared to normal cells. It details how genetic mutations and environmental factors, such as chemical carcinogens and viral infections, contribute to malignant transformations. The discussion also highlights the complex interplay between oncogenes, tumor suppressor genes, and the molecular pathways that regulate cell growth and differentiation.













































































