Download as PDF, PPTX


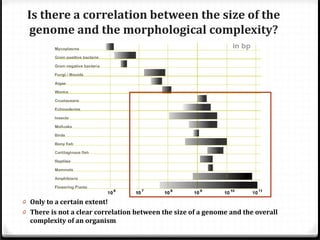
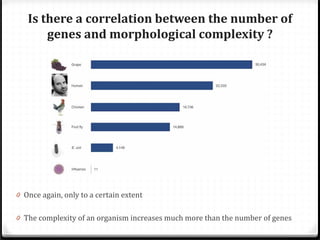
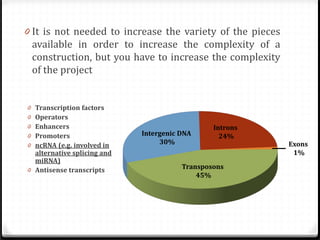
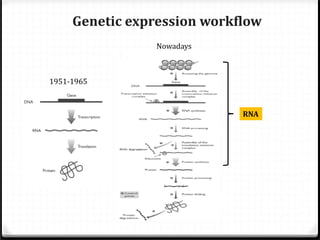
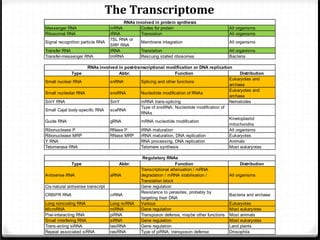

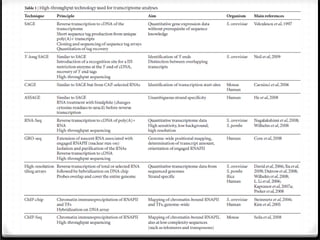
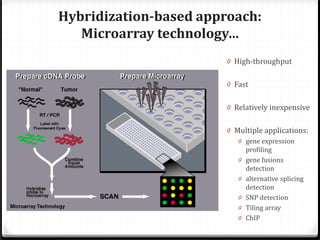



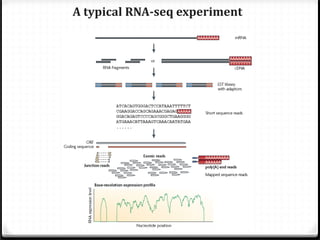
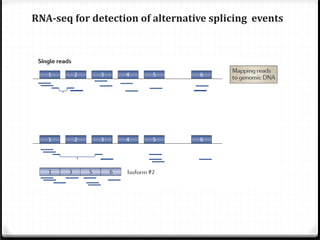
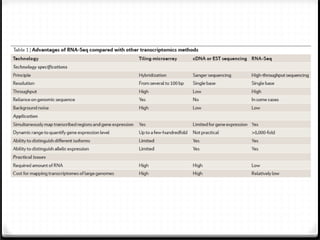




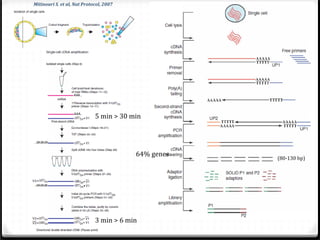
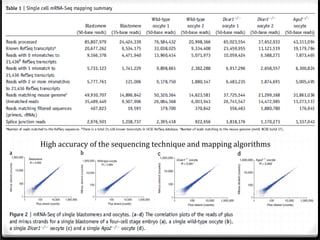




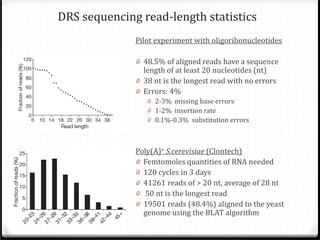
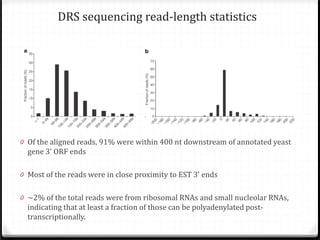




This document summarizes recent advances in transcriptome analysis technologies. It discusses limitations of microarray-based approaches and how next-generation sequencing-based RNA-seq provides more comprehensive transcriptome profiling. RNA-seq can detect thousands of new transcript variants and isoforms. It also describes direct RNA sequencing without cDNA conversion, revealing polyadenylation profiles with single-molecule resolution. Comprehensive polyadenylation maps in human and yeast showed previously unannotated sites and alternative polyadenylation, providing insights into regulatory mechanisms.























































































