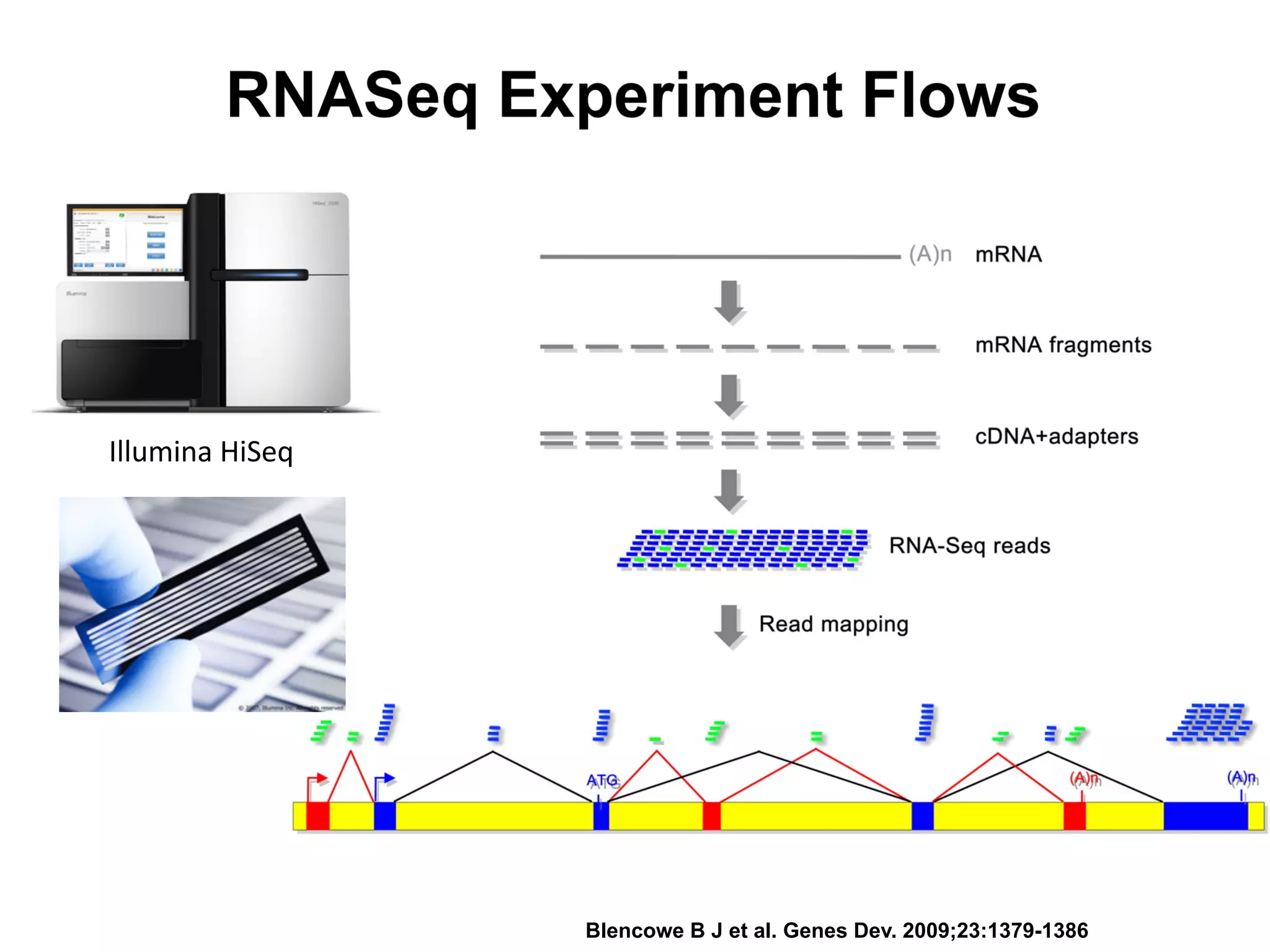
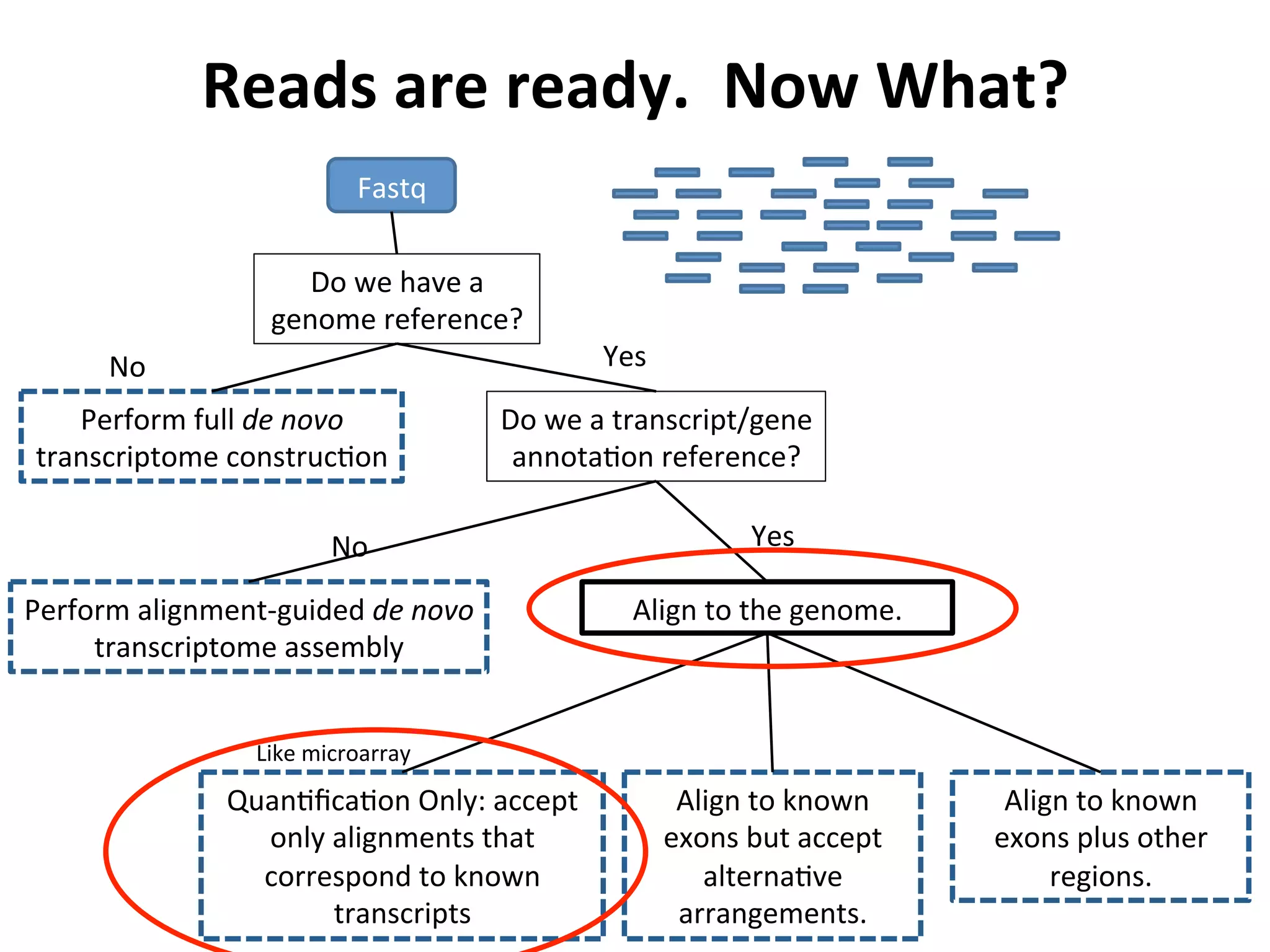
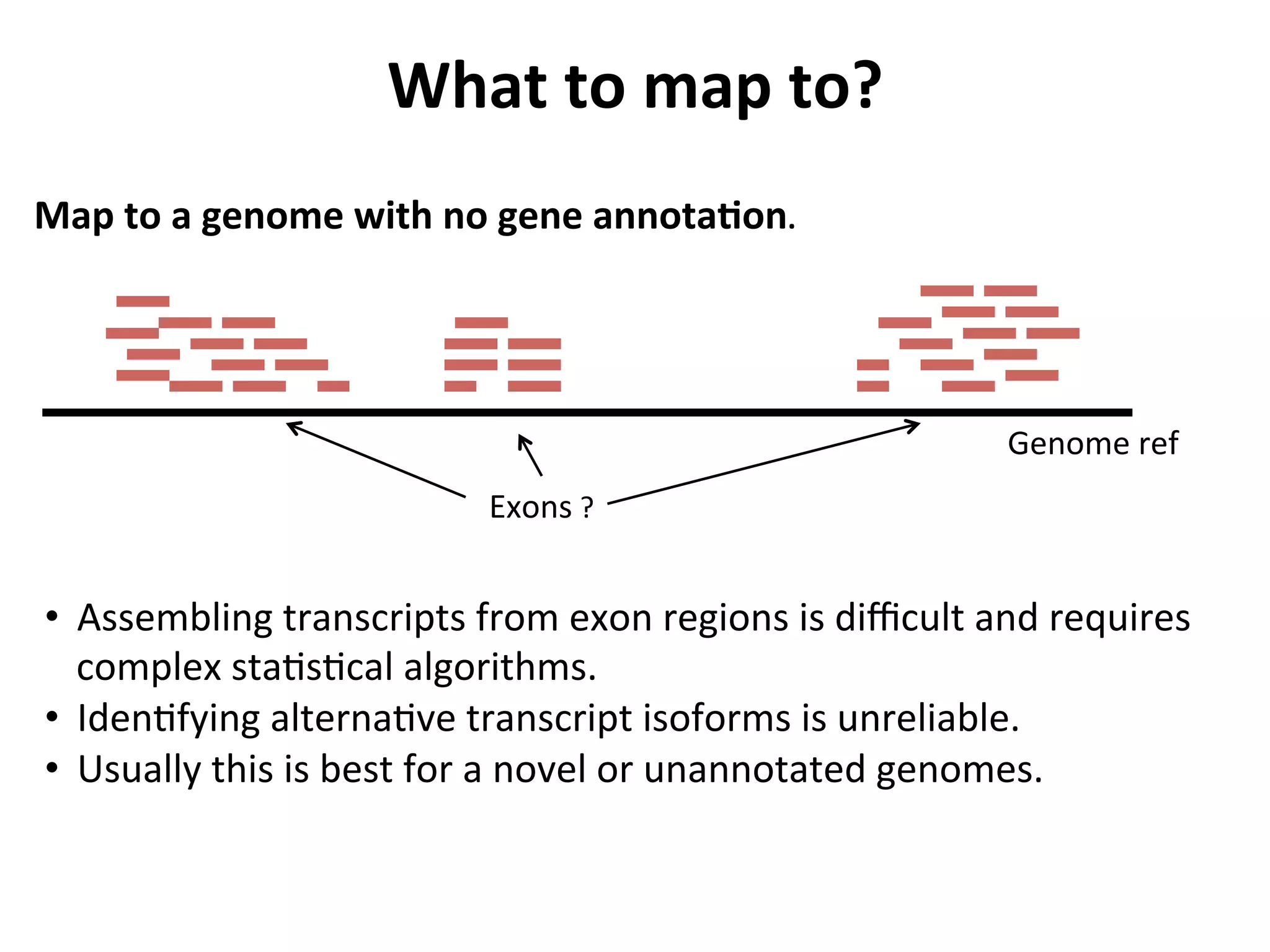
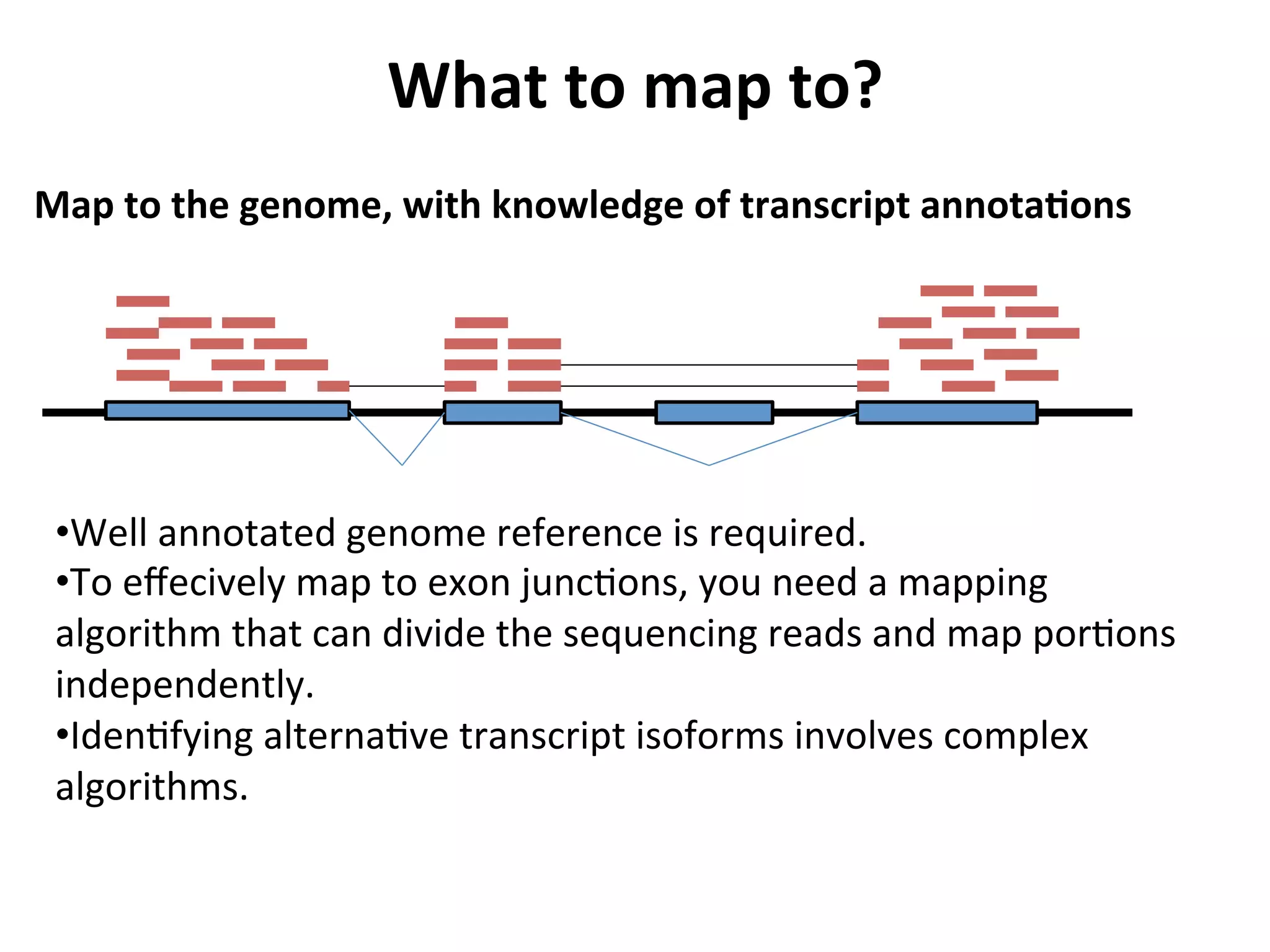
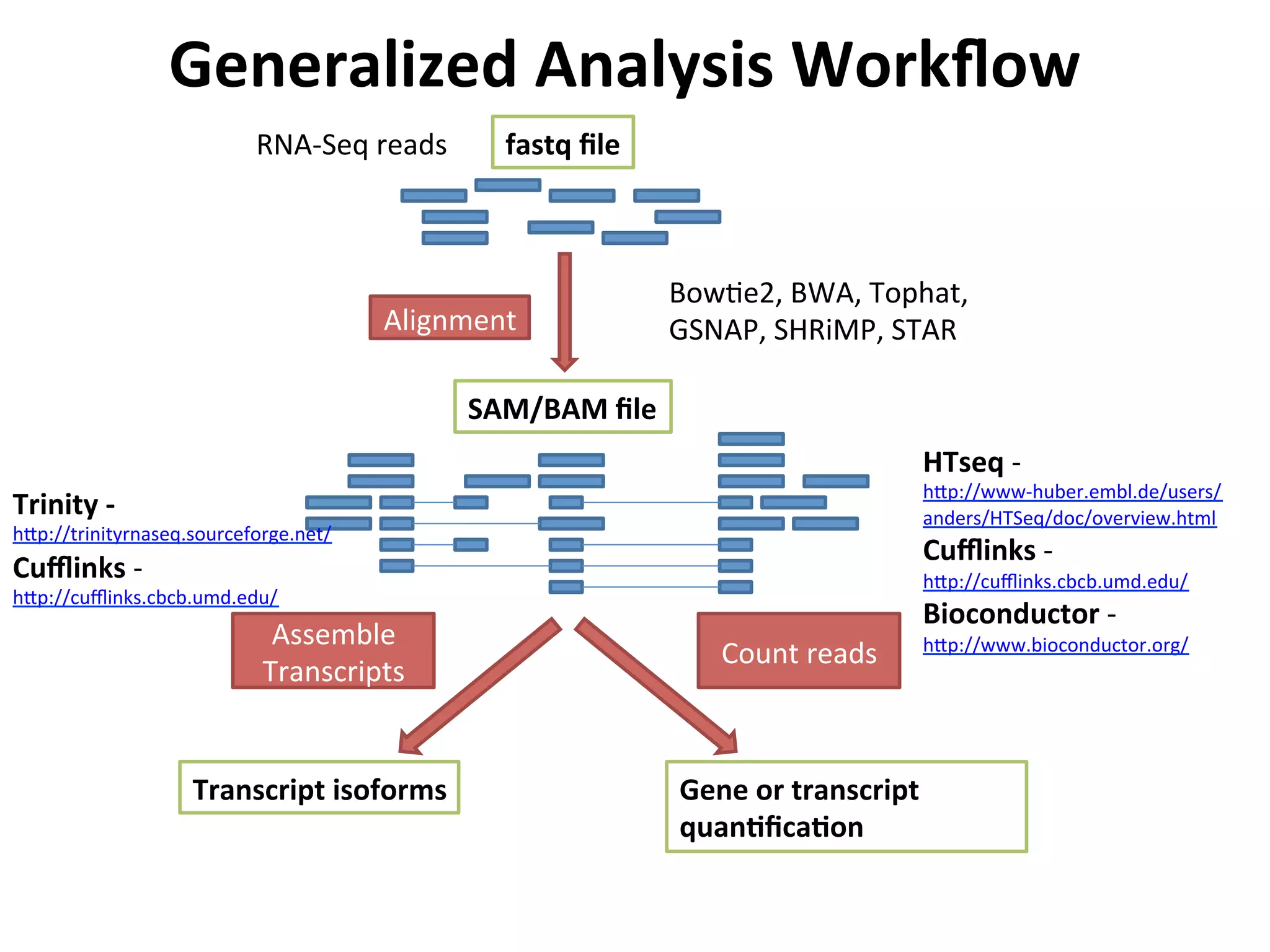
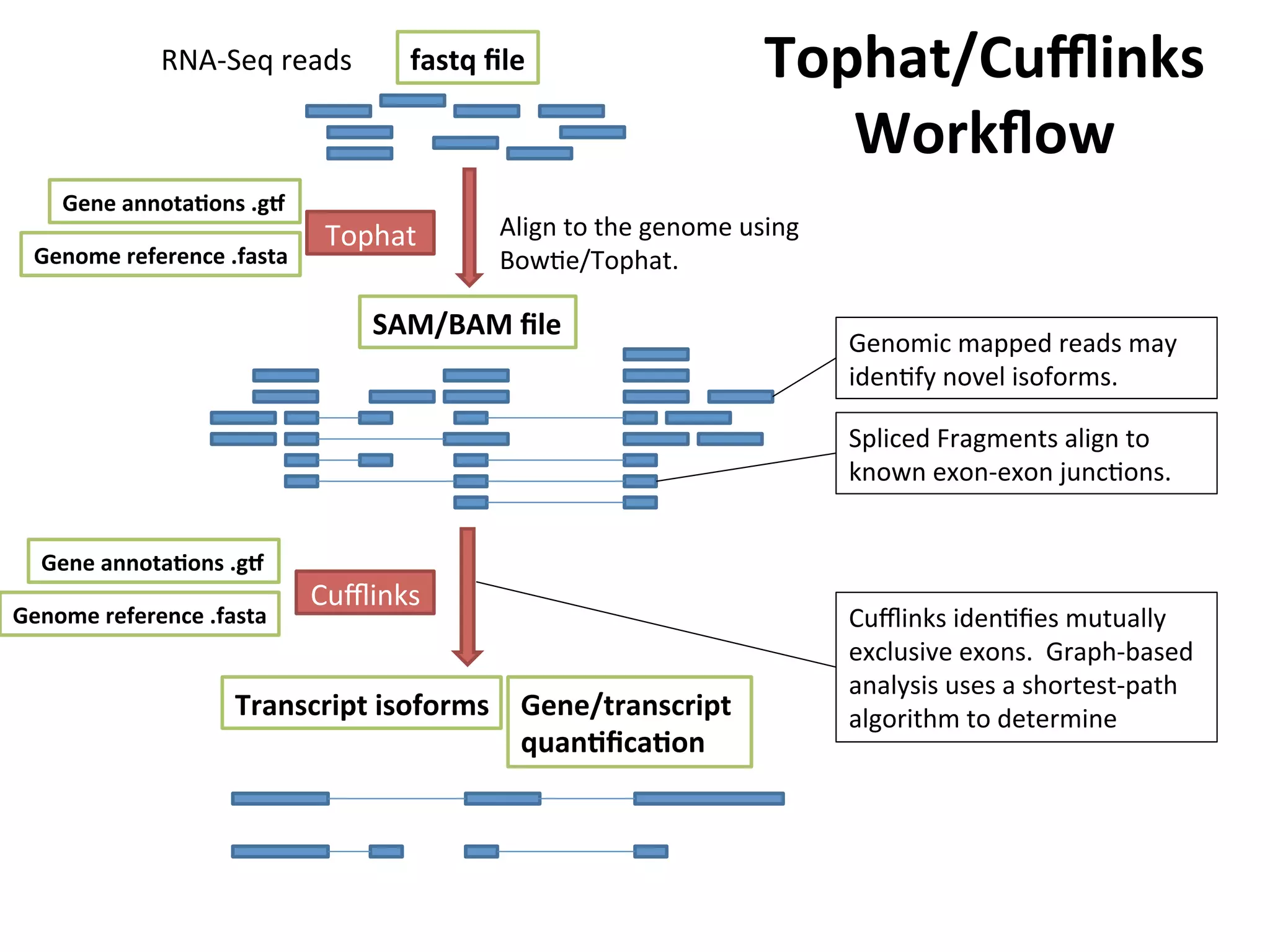
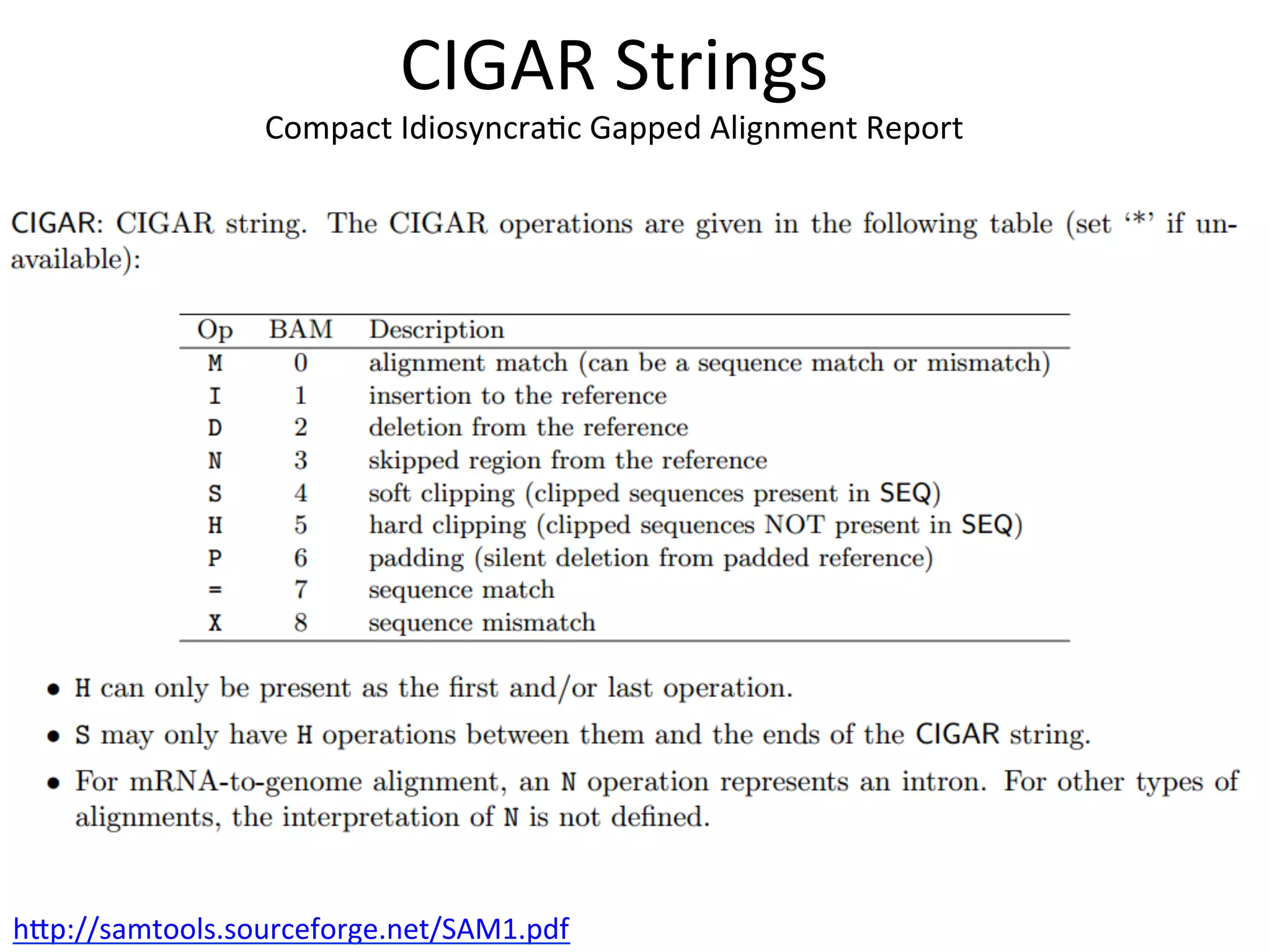
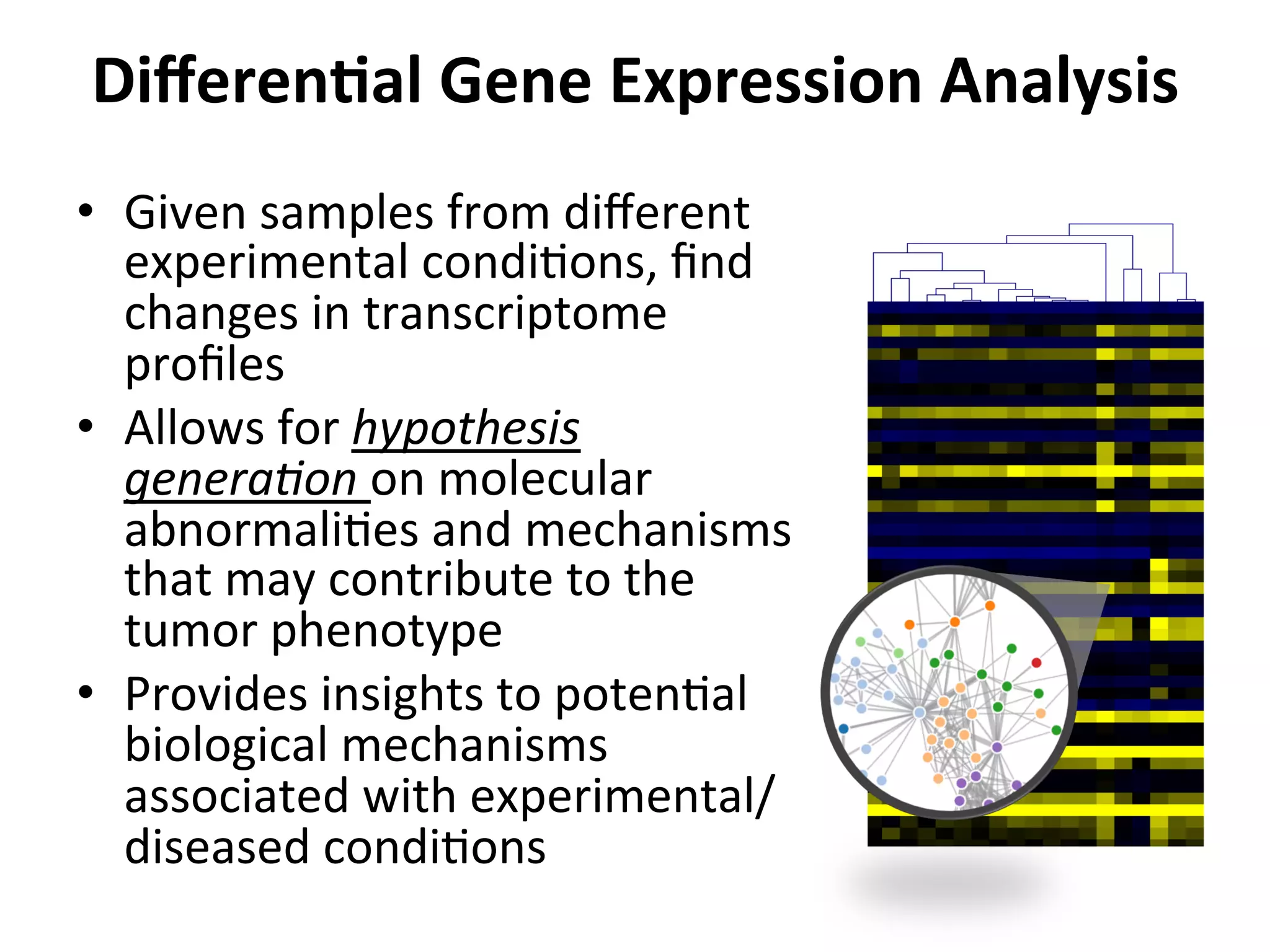
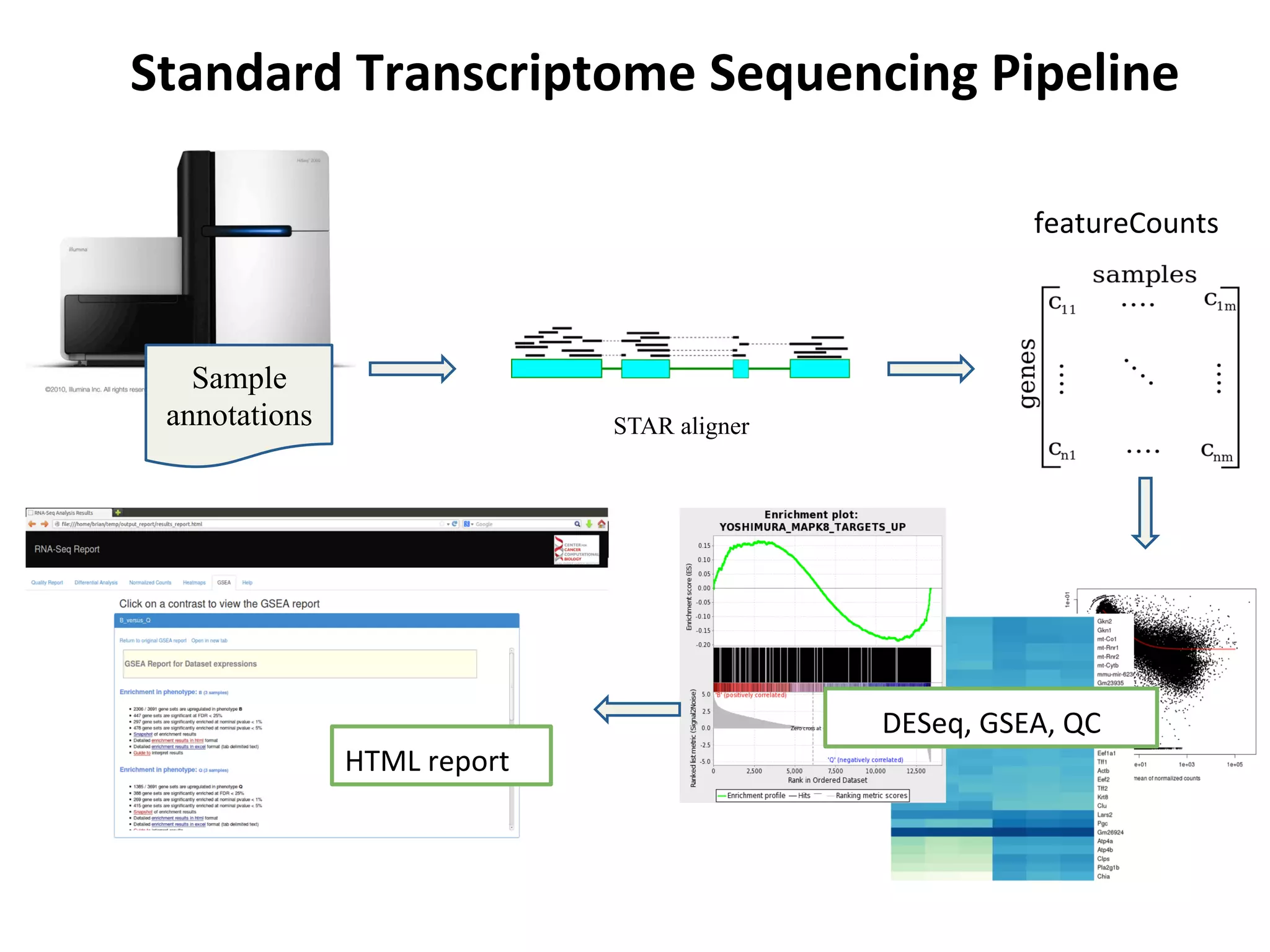
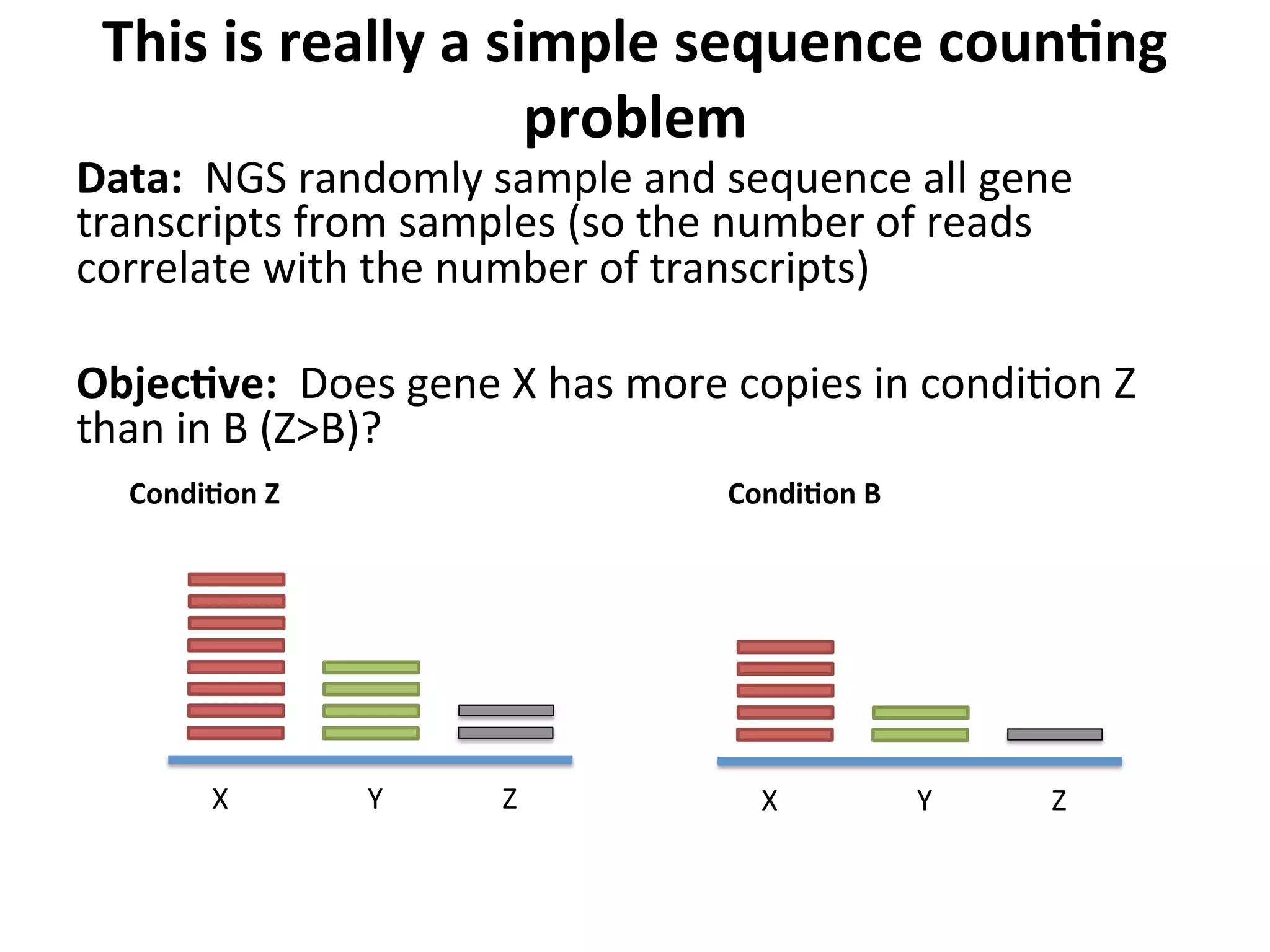
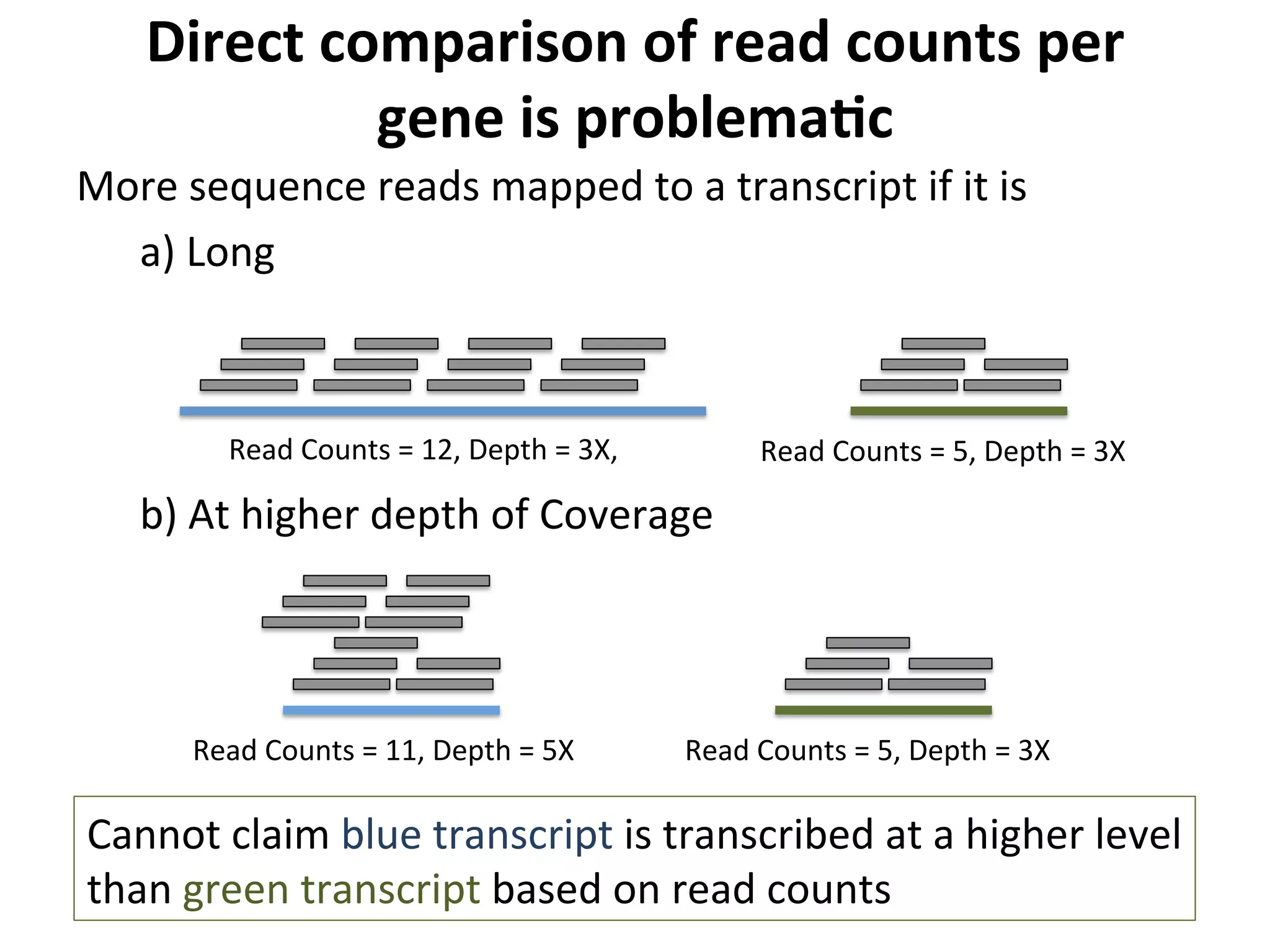
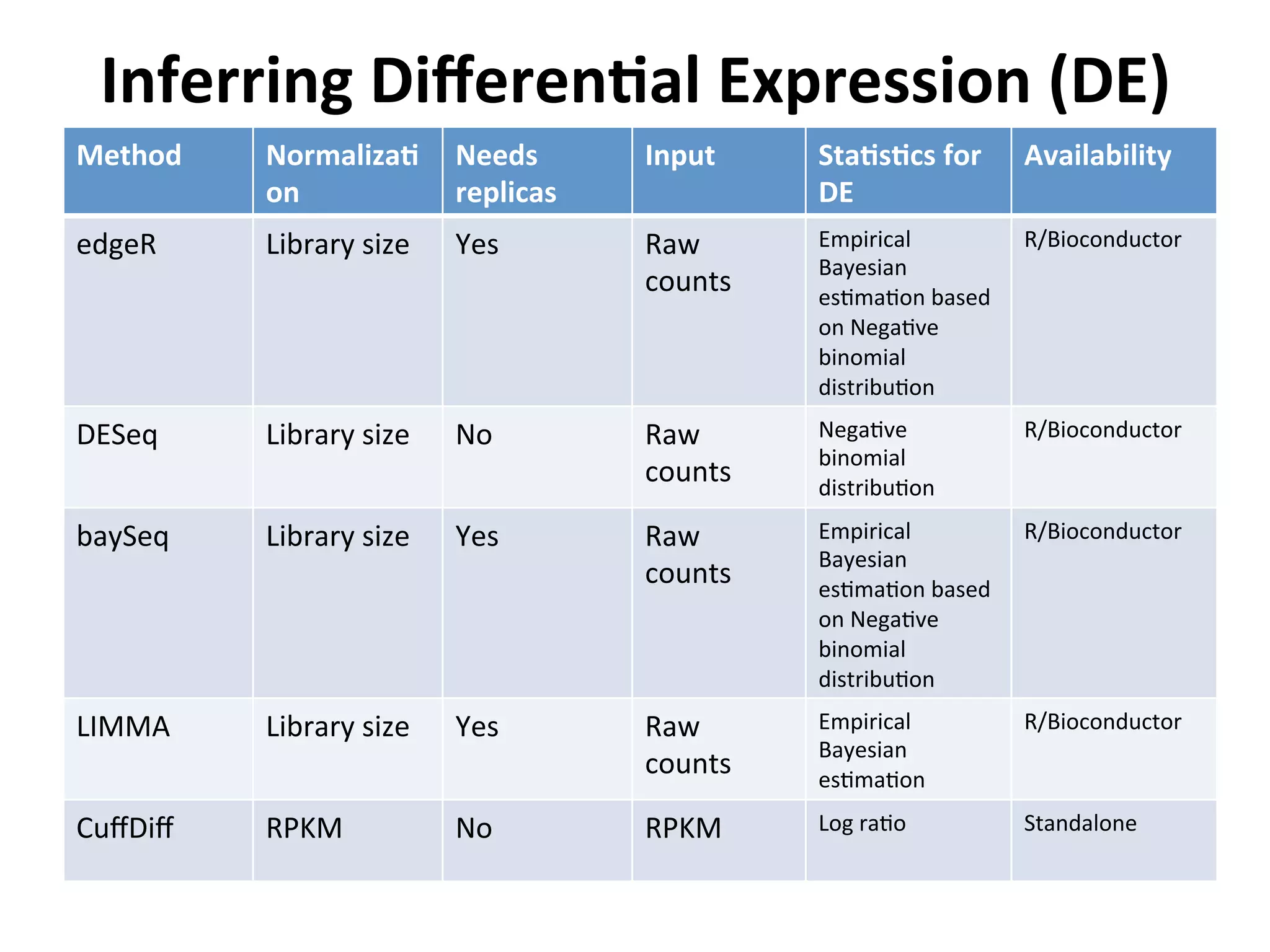
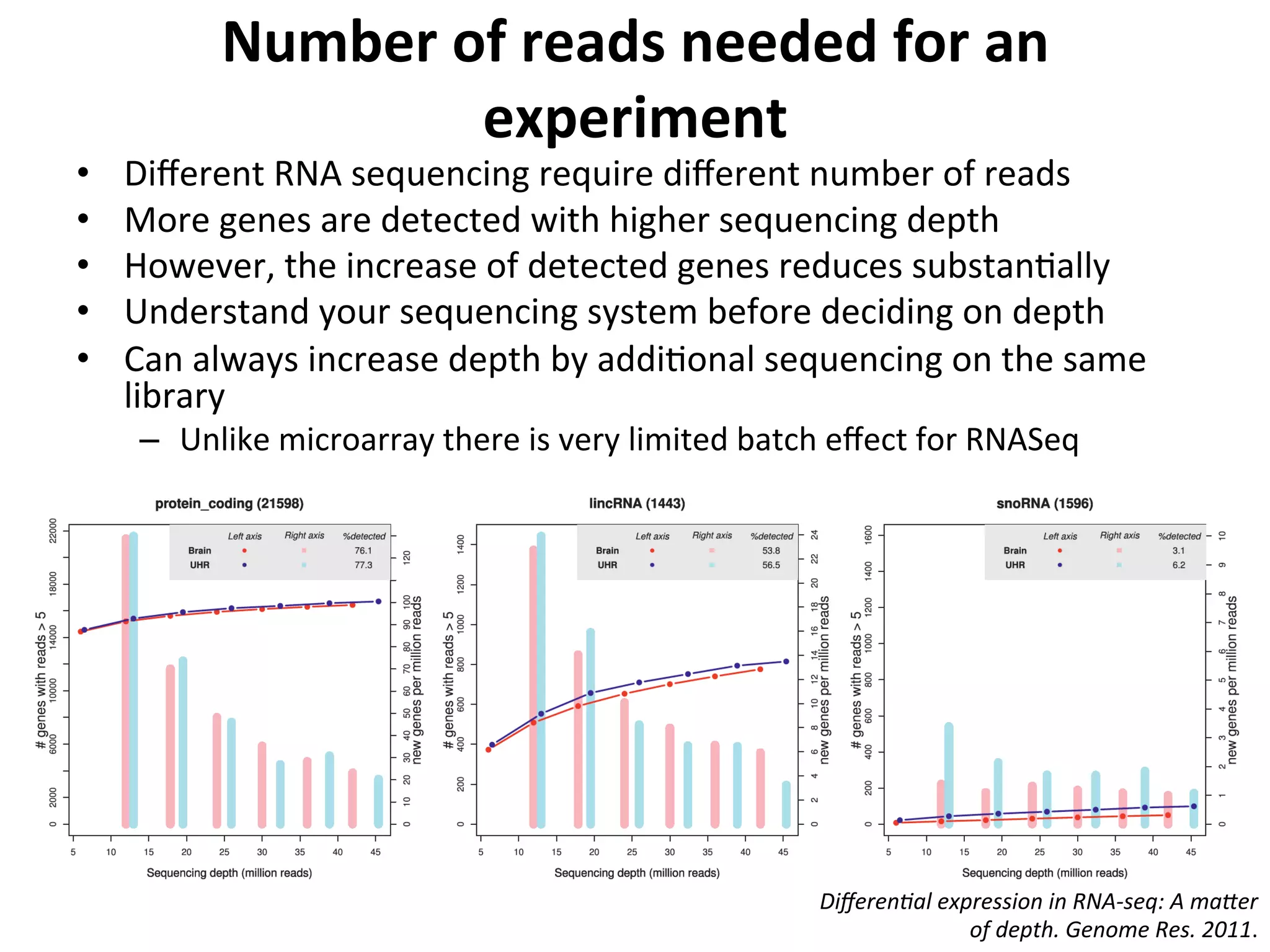
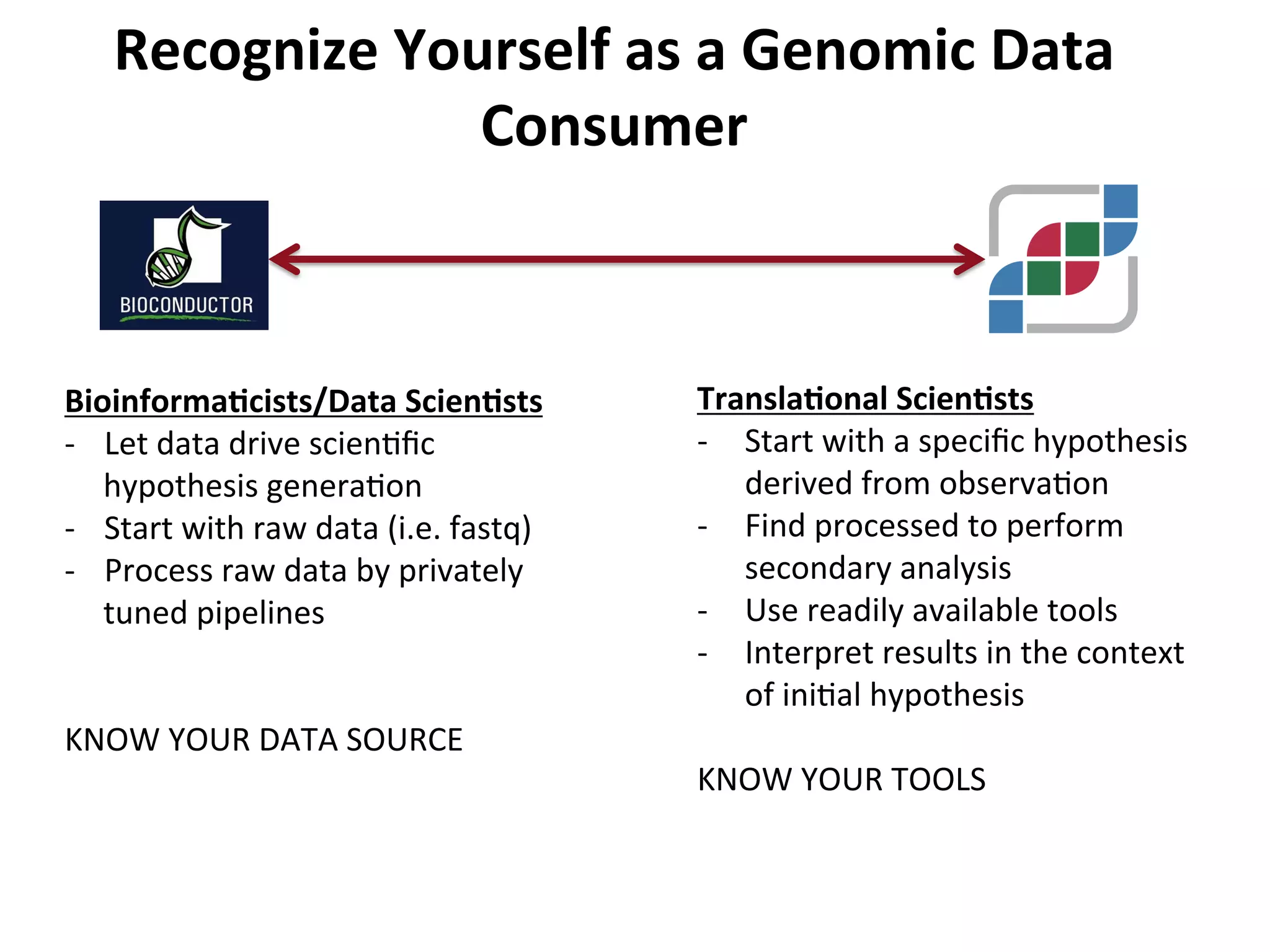
This document provides an overview of RNAseq analysis workflows. It discusses preparing raw sequencing reads, aligning reads to a reference genome or transcriptome, and using tools like Tophat and Cufflinks to assemble transcripts and quantify gene and transcript expression. Key steps include mapping reads, assembling transcripts, quantifying expression at the gene or transcript level, and using the results to identify differentially expressed genes between experimental conditions.
















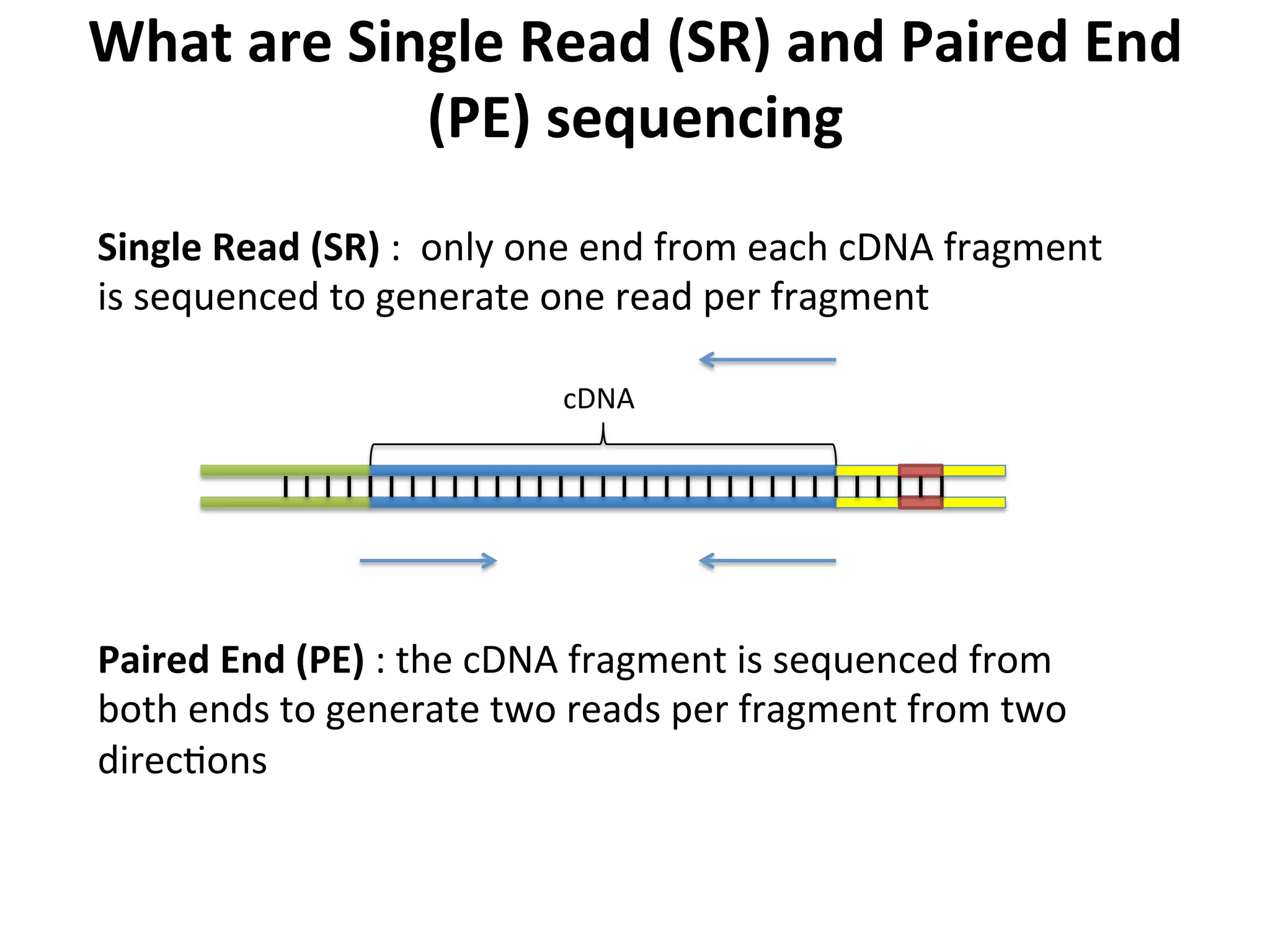
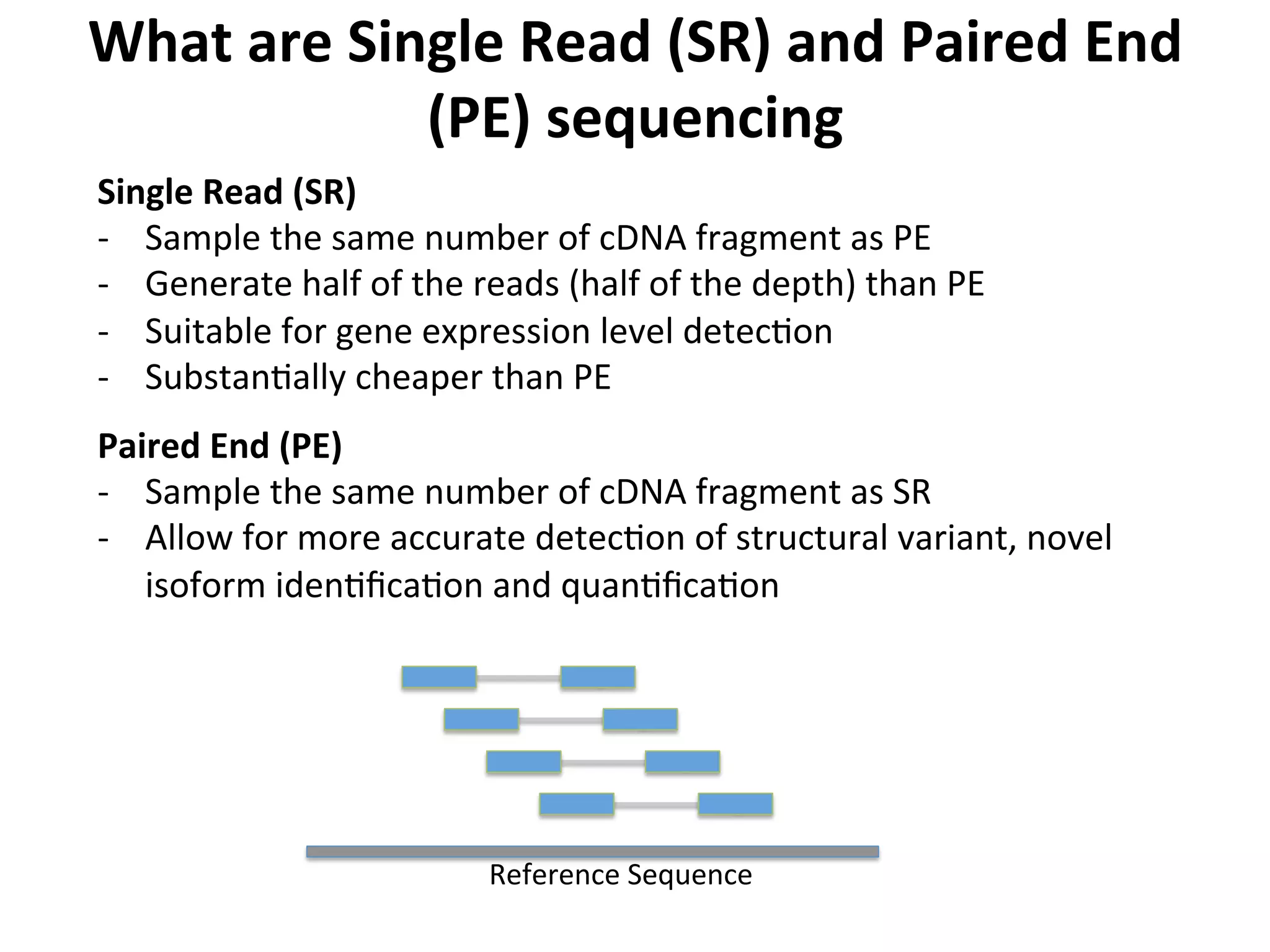
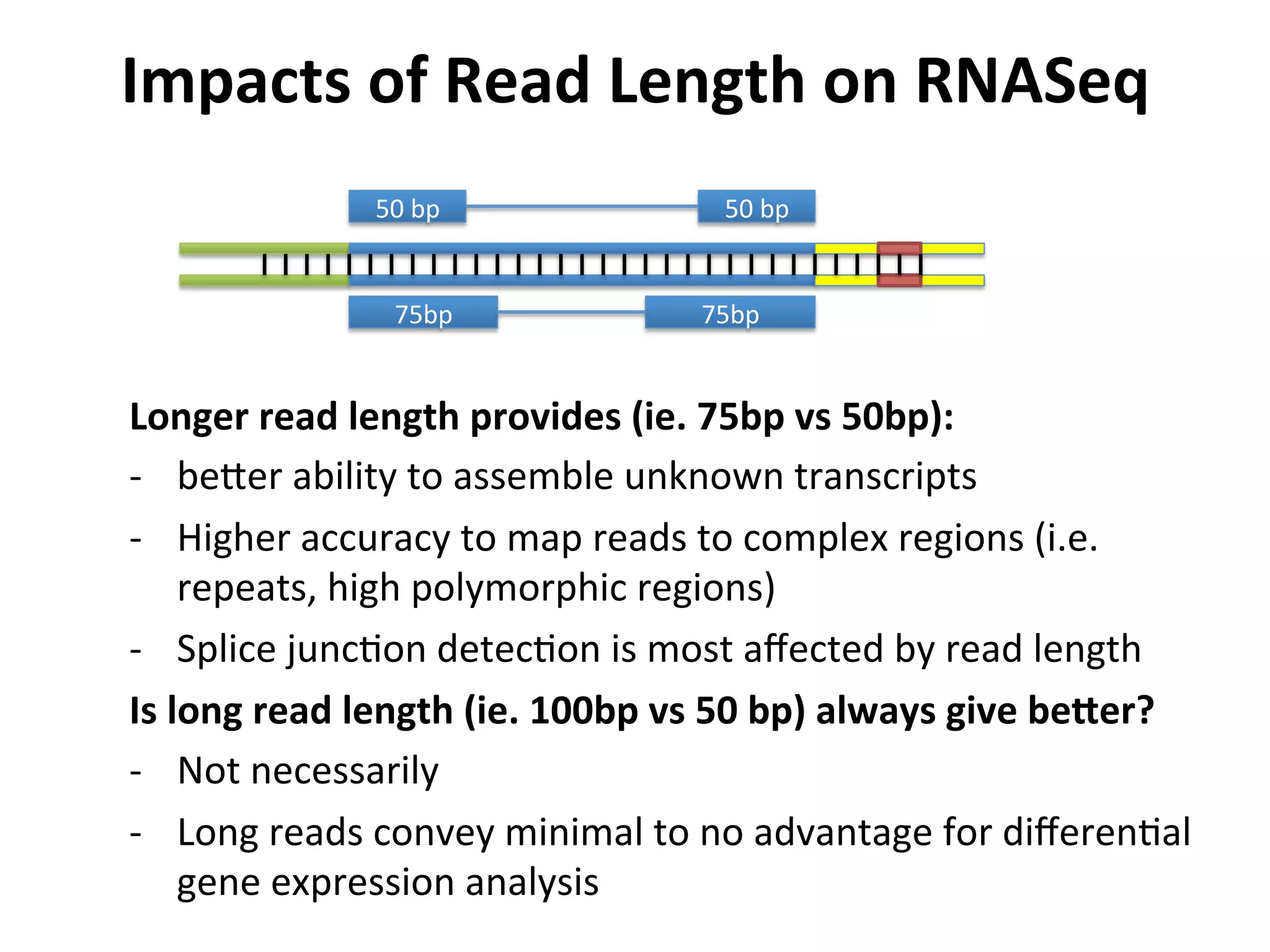
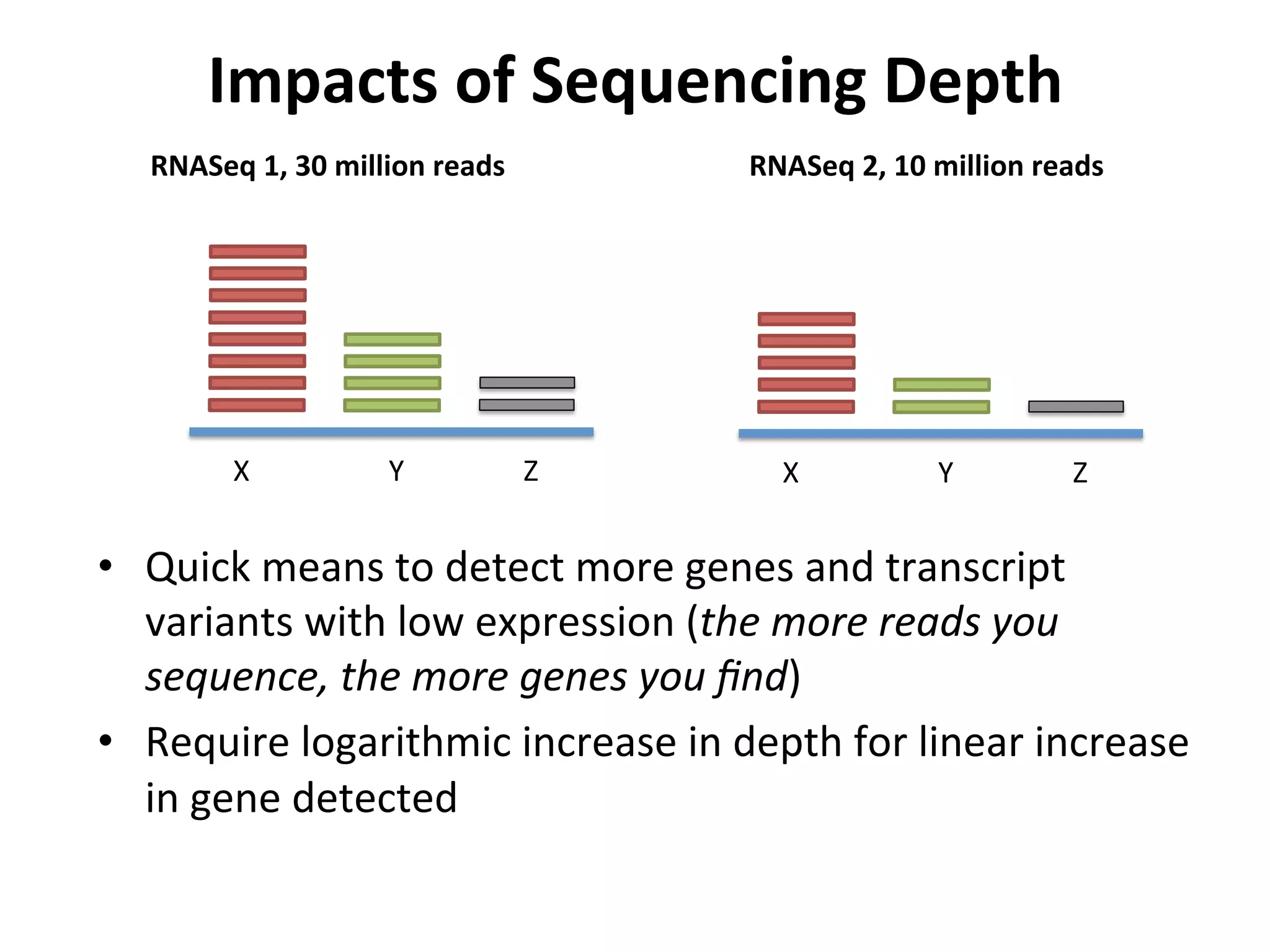











































































![Vibe Coding vs. Spec-Driven Development [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/vibecodingvsspecdrivendevelopment-251209105622-43f455e7-thumbnail.jpg?width=640&height=640&fit=bounds)















