Downloaded 56 times








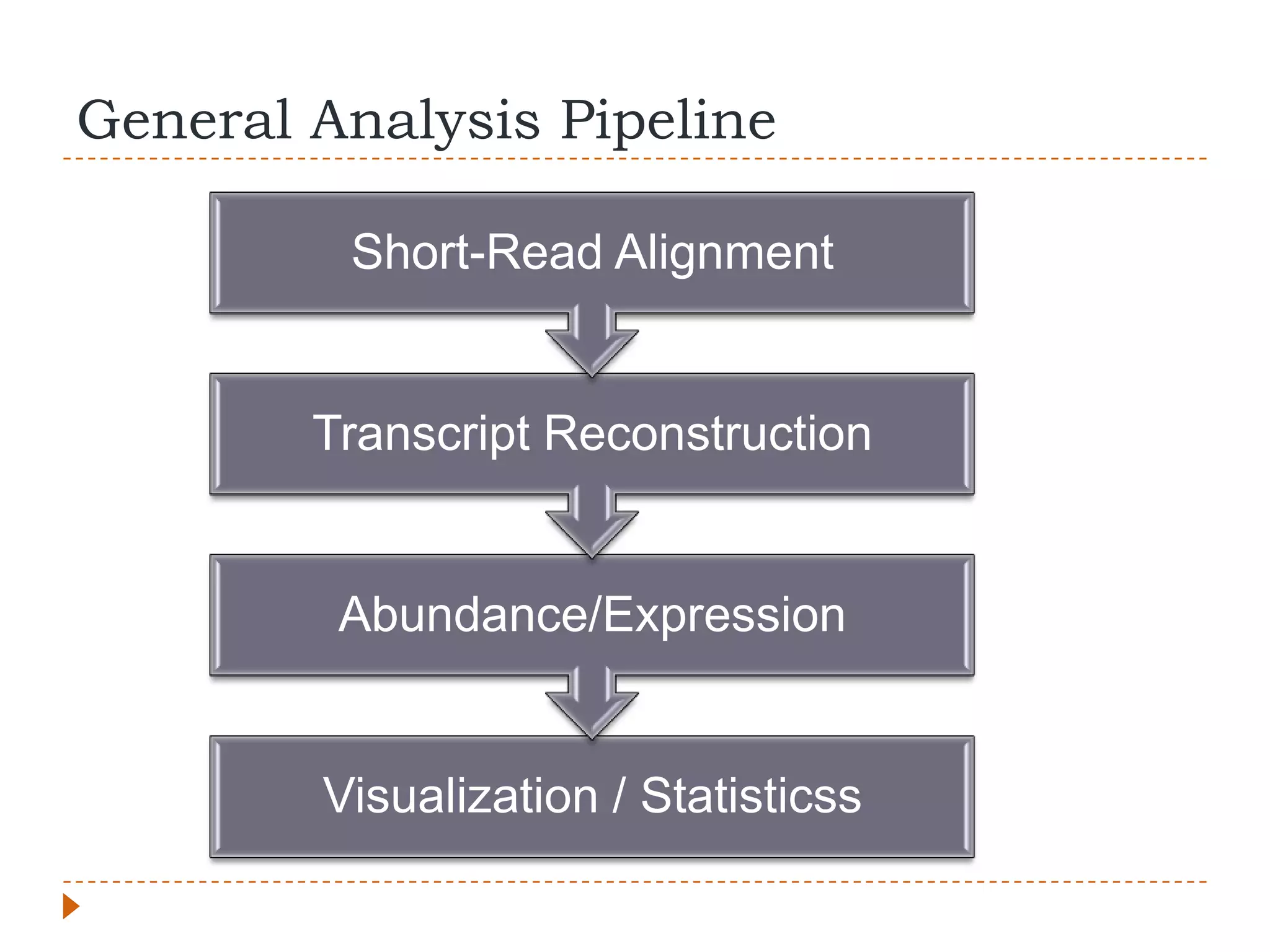
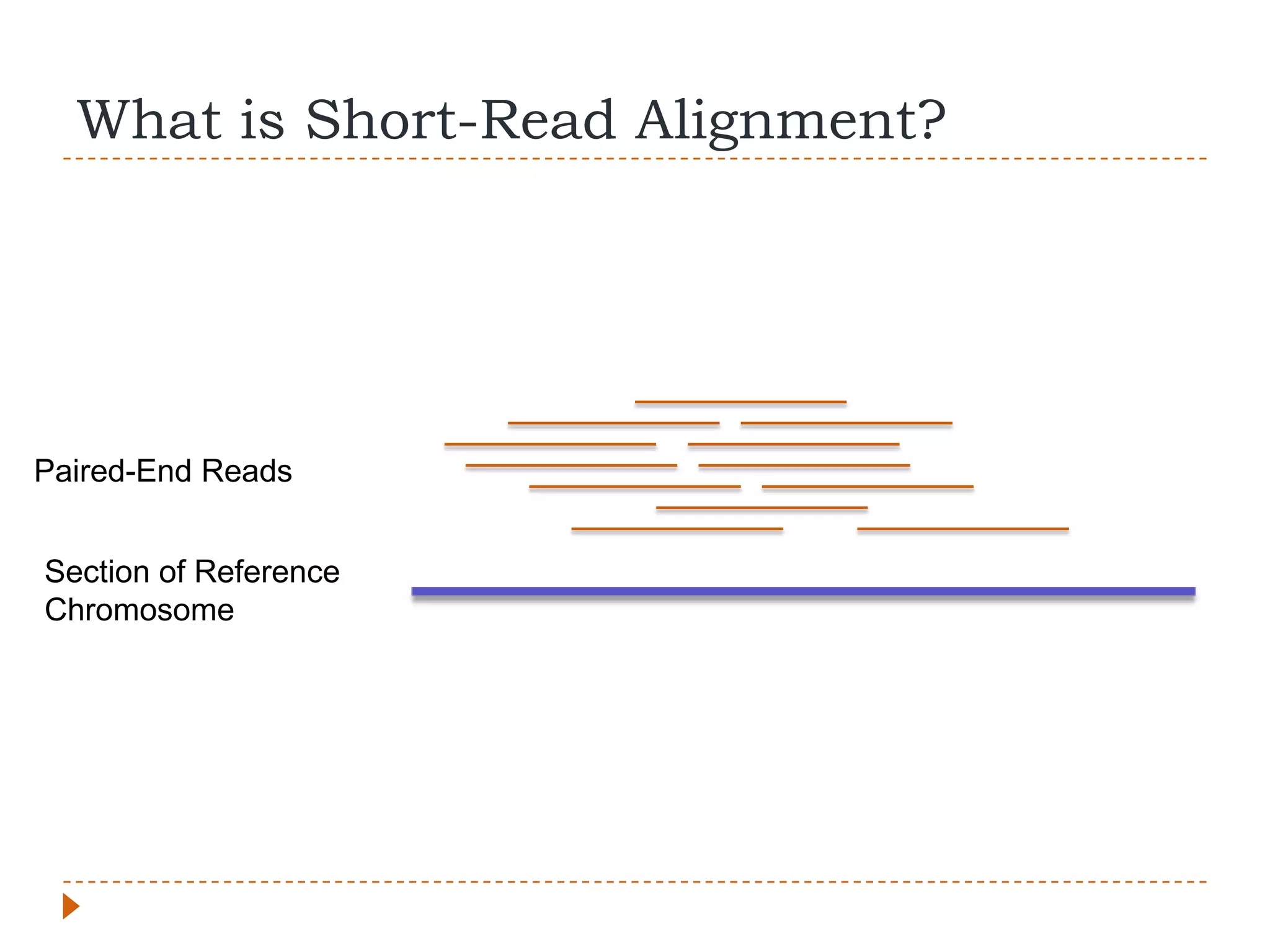

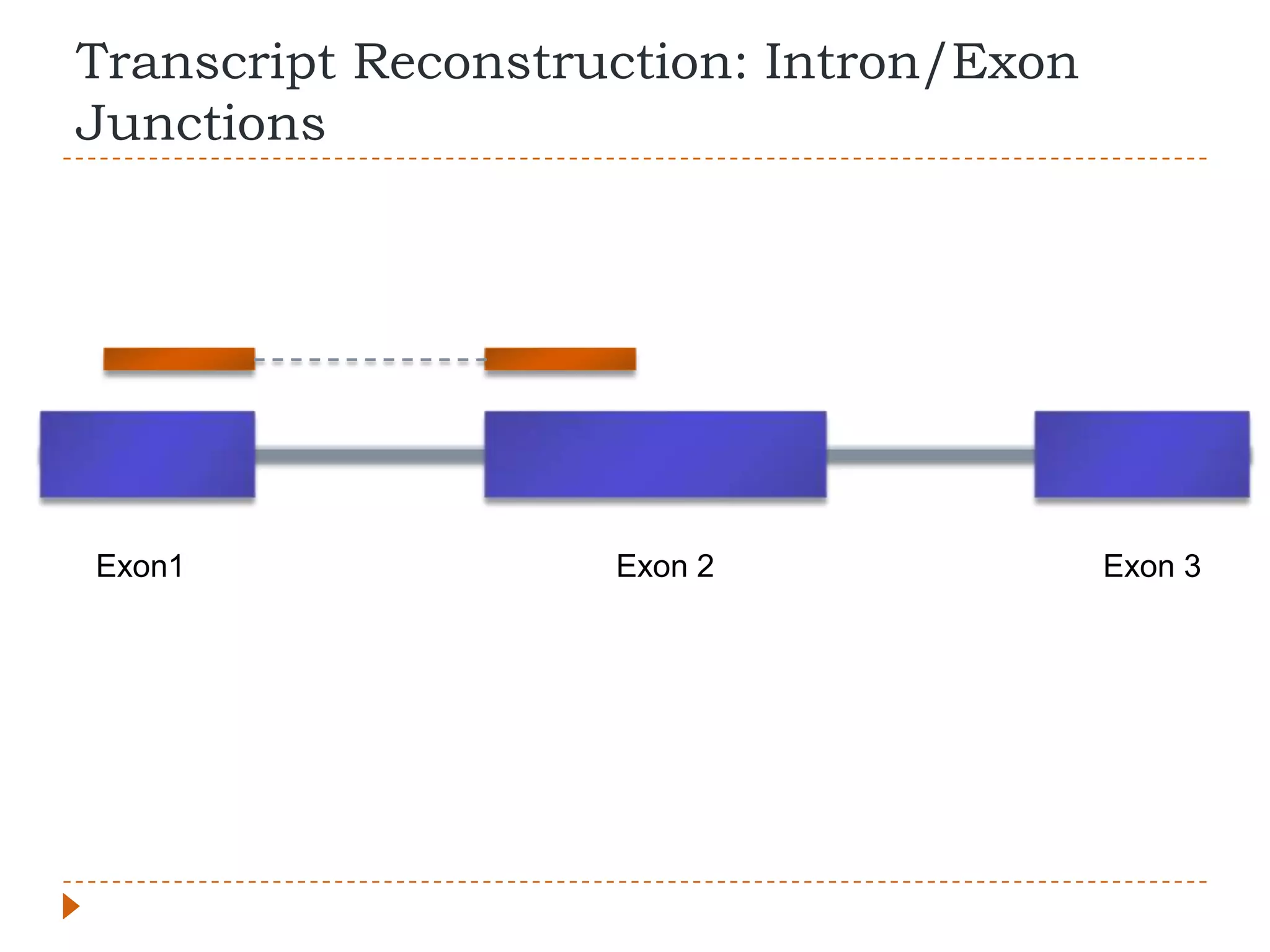
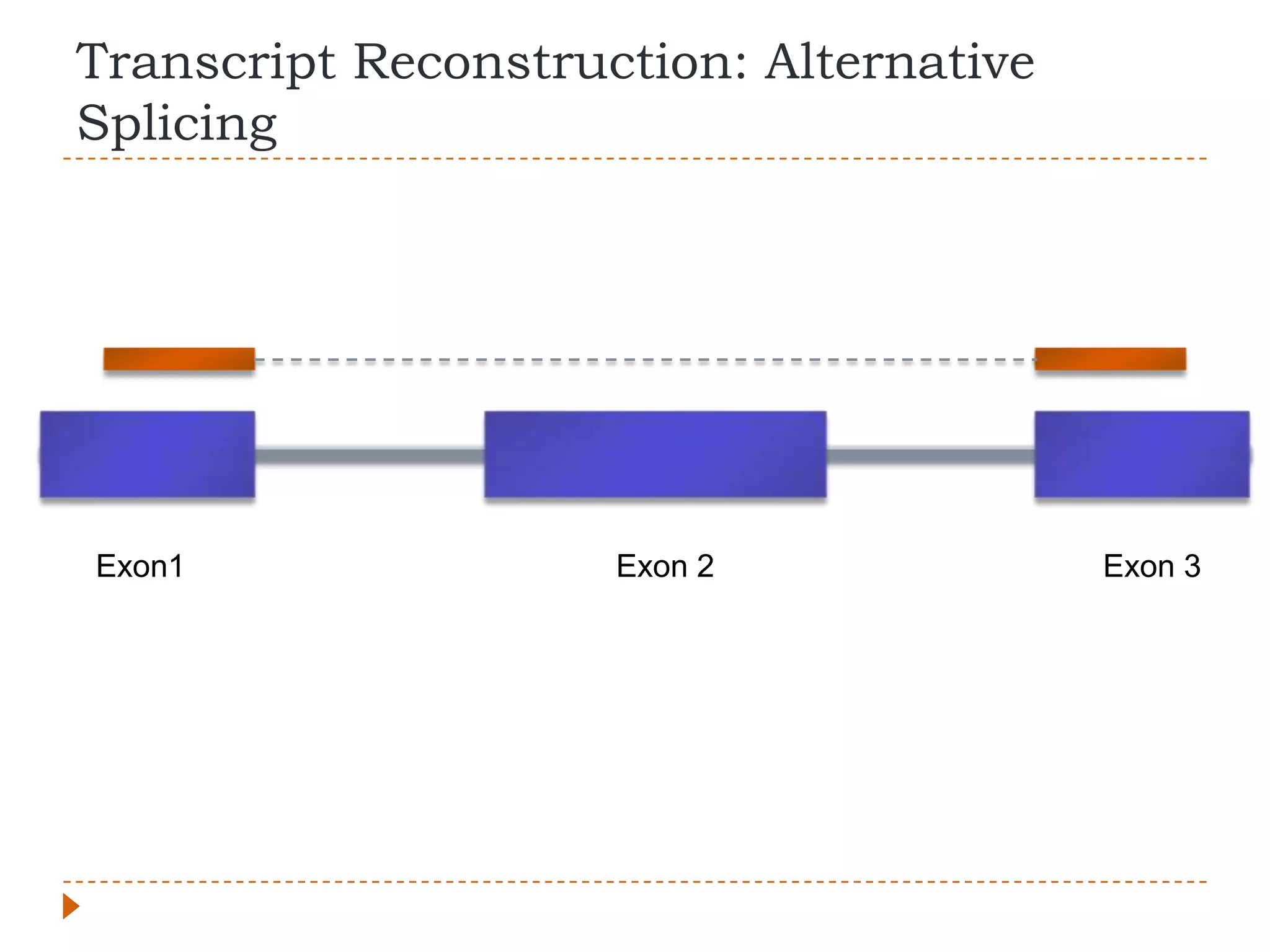
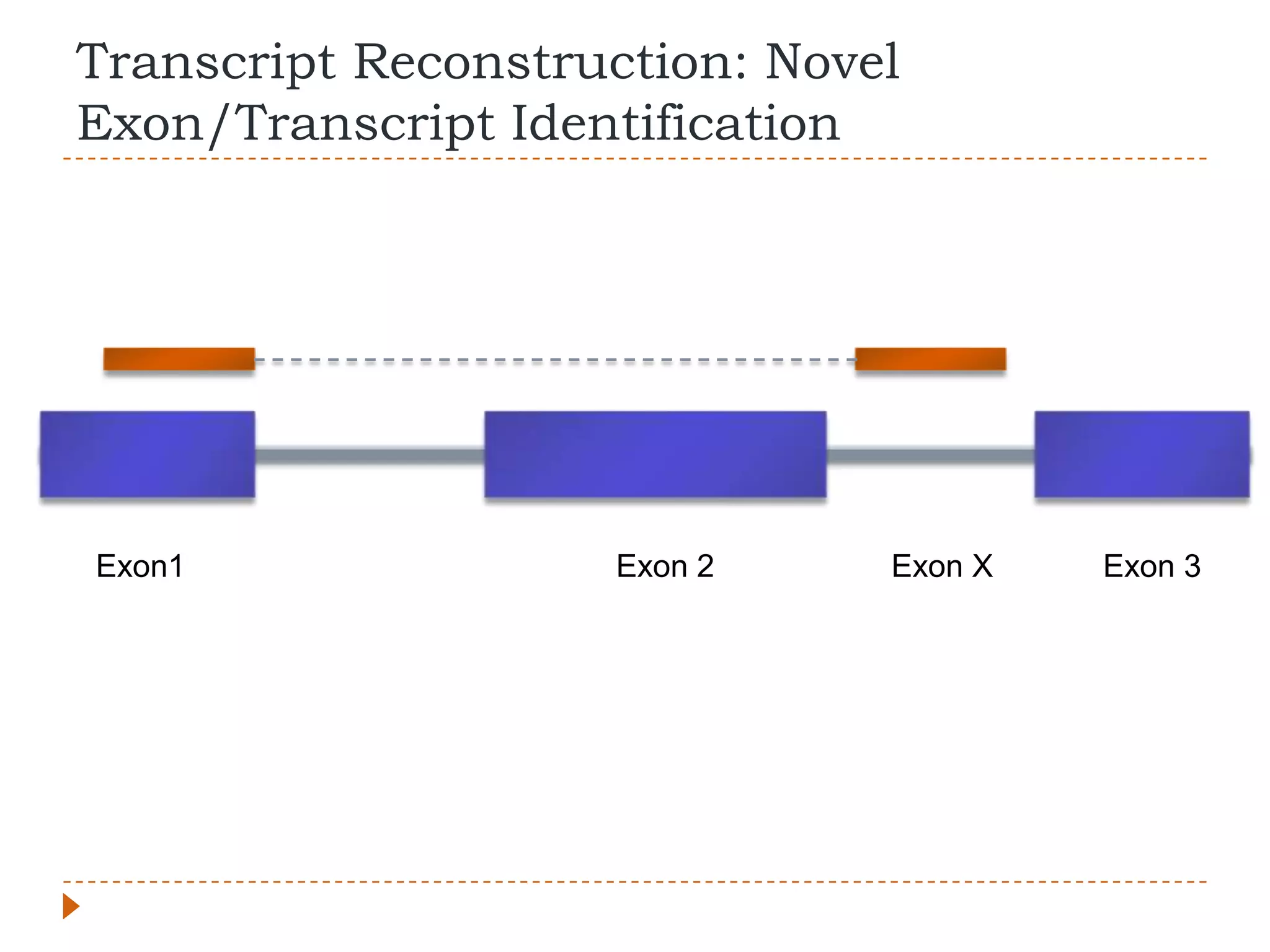
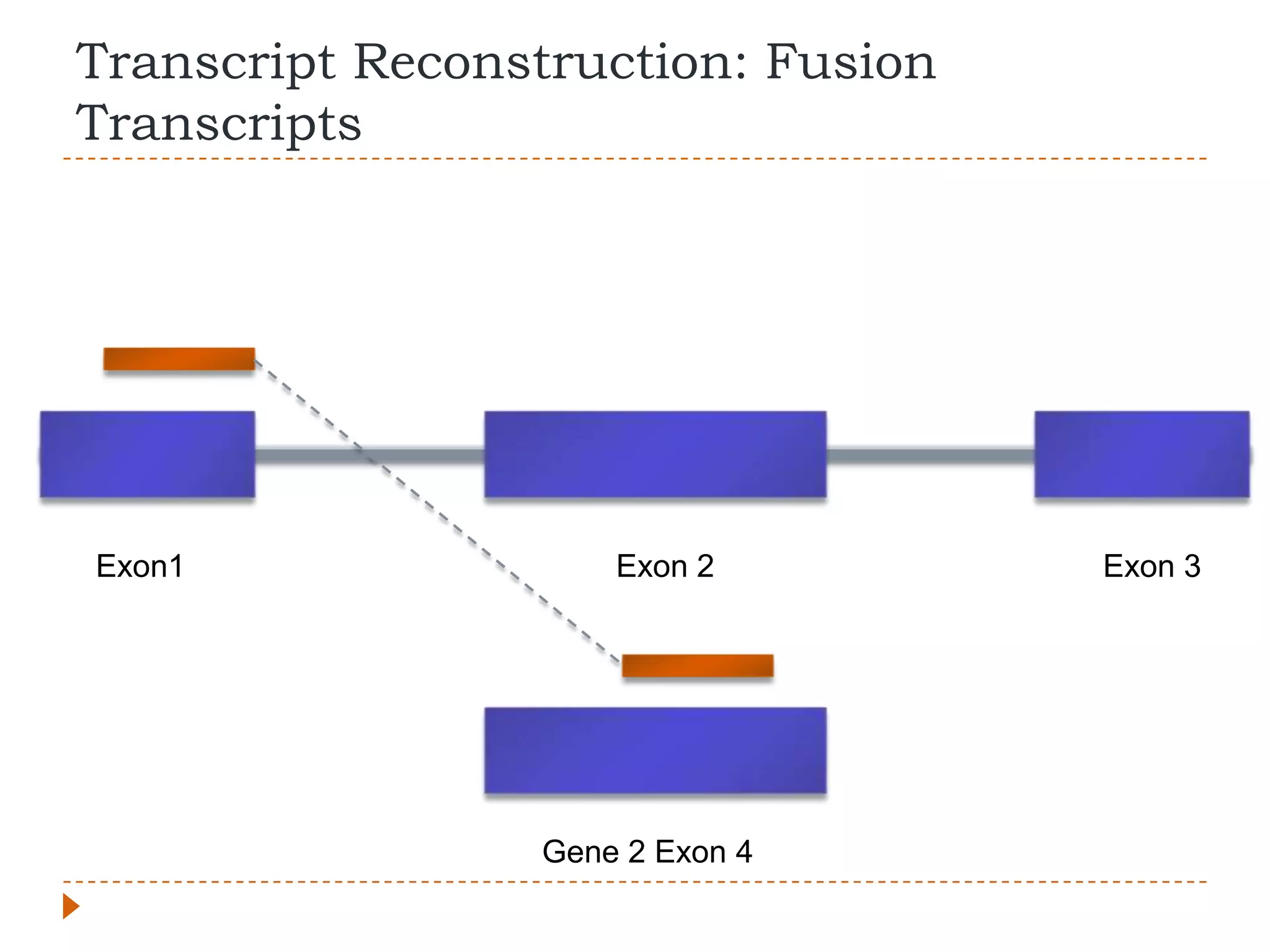
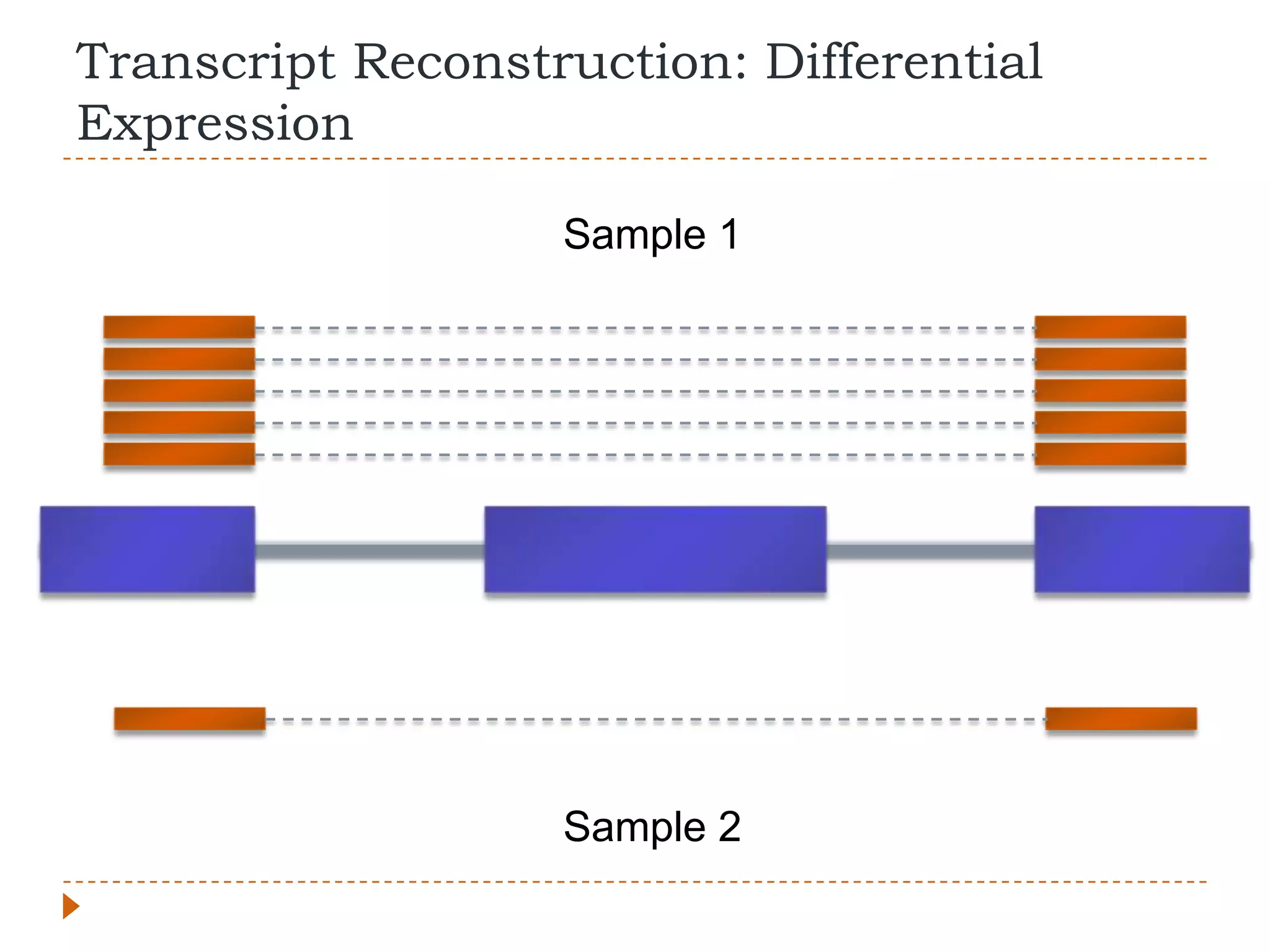


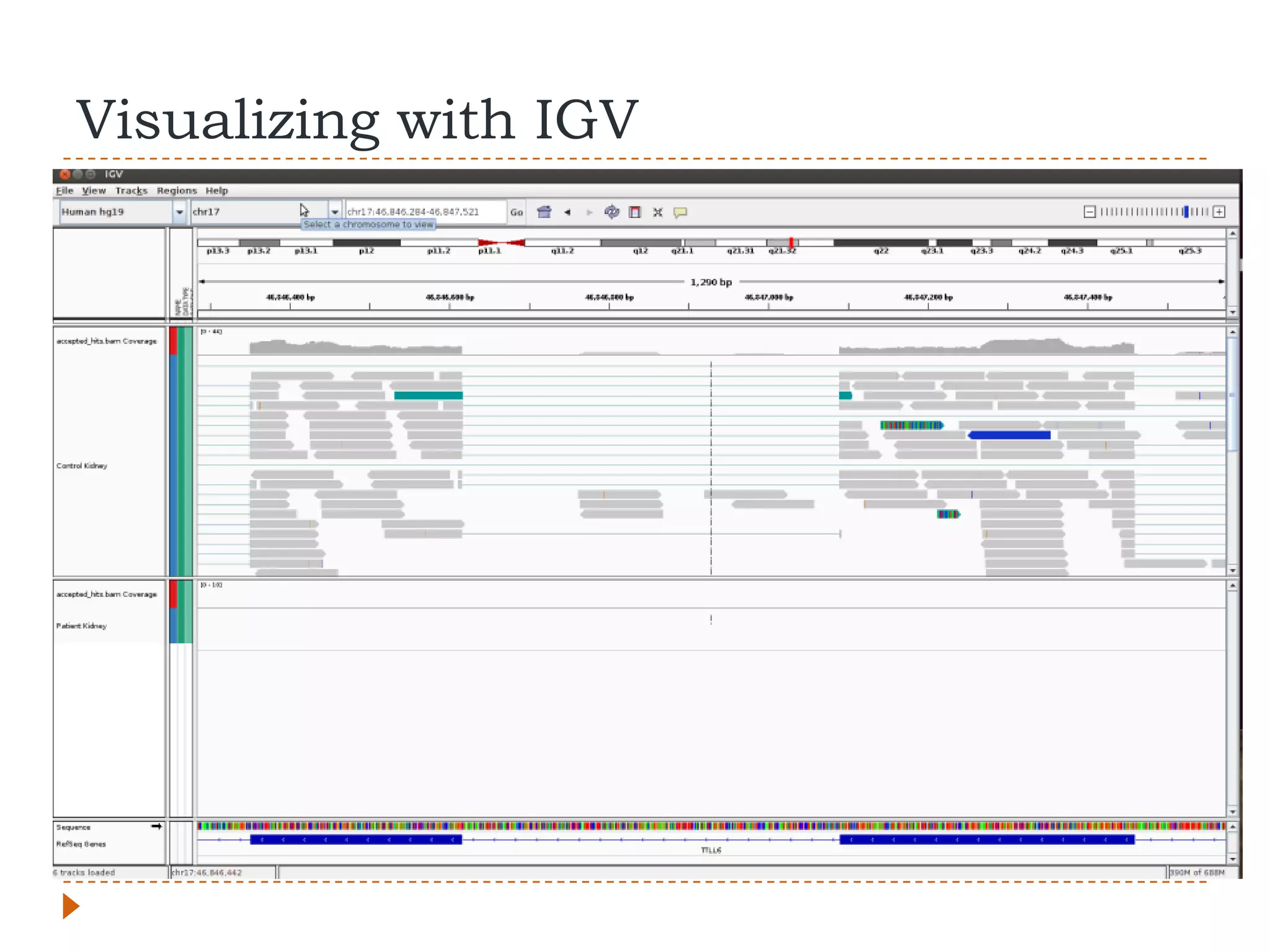
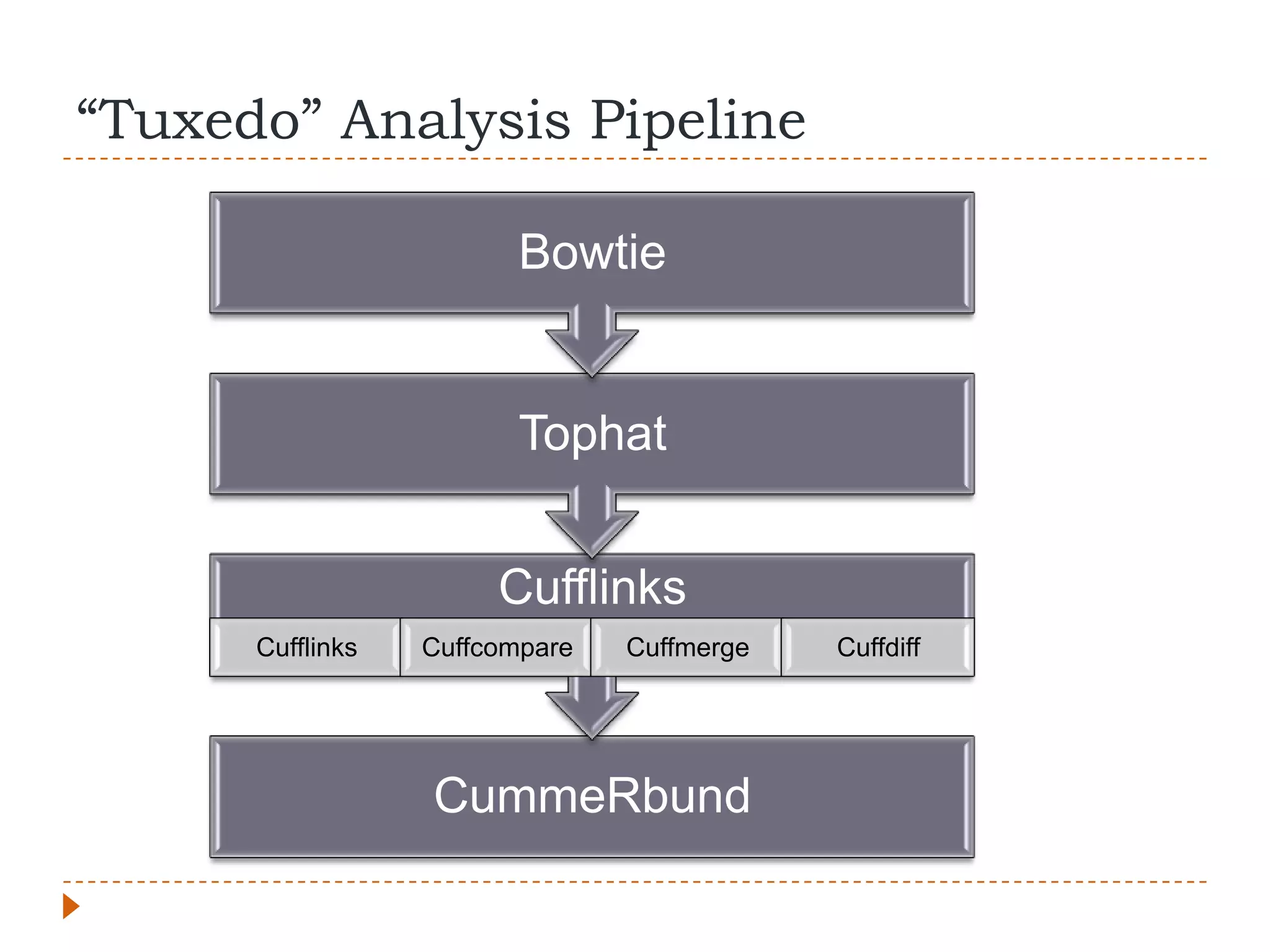









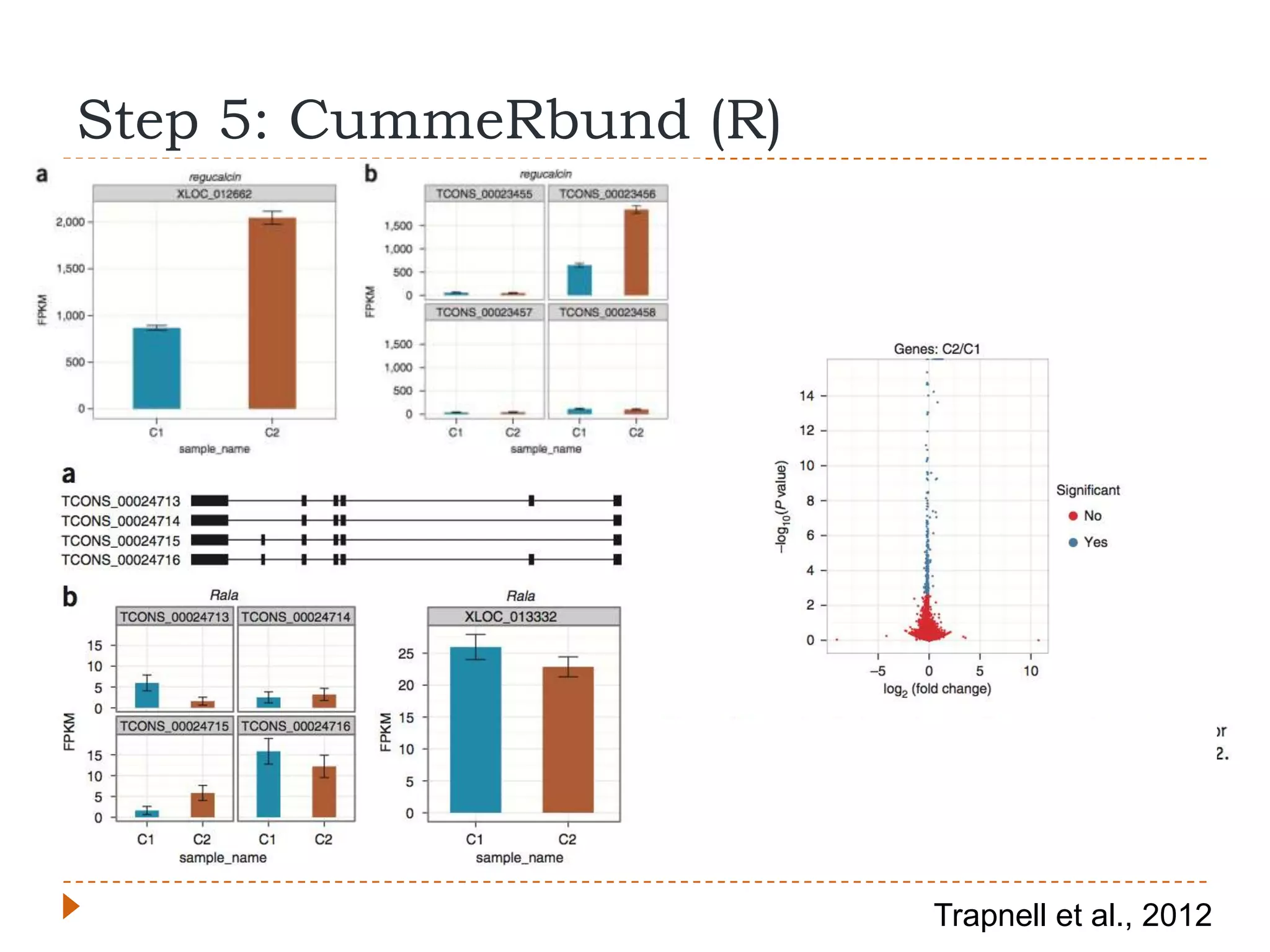
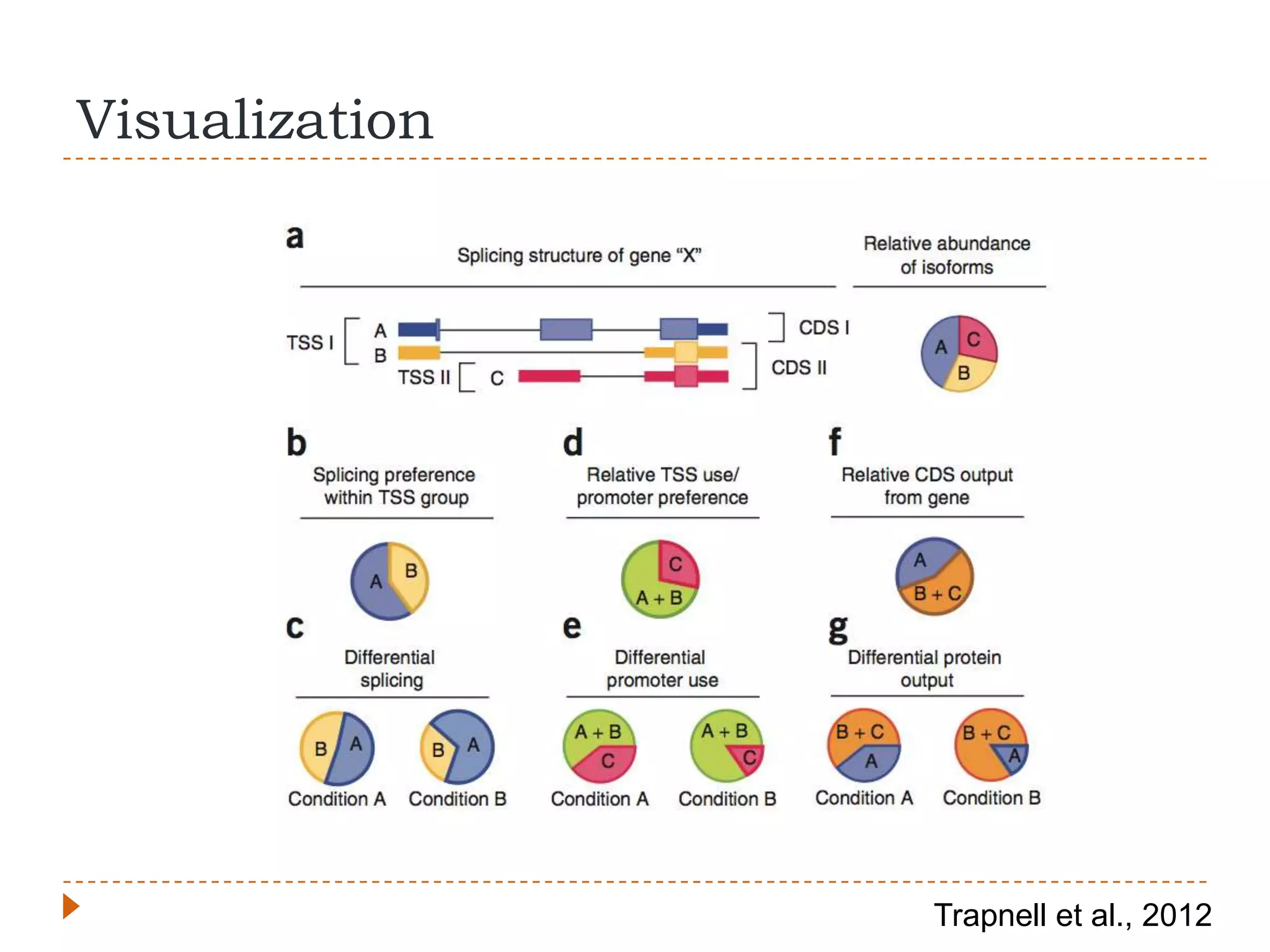
















This document provides an overview of RNA-Seq analysis. It begins with considerations for RNA-Seq experiments such as computational requirements. It then describes the general RNA-Seq analysis workflow including short-read alignment, transcript reconstruction, abundance estimation, visualization, and statistics. The document focuses on explaining the "Tuxedo" analysis pipeline which includes Bowtie, Tophat, Cufflinks, Cuffmerge, Cuffdiff and CummeRbund. It provides examples of commands for each step and discusses alternative tools. The document concludes with resources for further information on RNA-Seq analysis.














































































