Download as PDF, PPTX

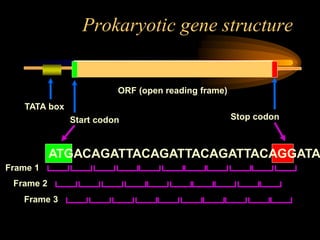








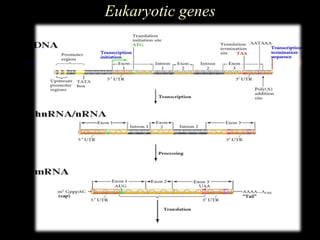







The document discusses gene prediction methods for prokaryotic and eukaryotic genes, highlighting the advantages and disadvantages of each approach. It details various gene finding strategies including rule-based, content-based, similarity-based, pattern-based, and ab-initio methods, as well as the use of machine learning techniques. Additionally, it reviews specific gene prediction programs and emphasizes the importance of combining multiple methods for improved accuracy.















































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)







